GO-Elite Analysis in AltAnalyze
AltAnalyze provides a powerful tool for performing enrichment analyses across a large array of biological categories to discover important functional relationships in your data as well as putative regulatory relationships (e.g., transcriptional, microRNA, cell-type interactions). This is done using the integrated software called GO-Elite. Several useful graphical outputs are produced from these analyses including heatmaps, networks and colored pathways based on these enriched terms and genes/metabolites. In addition to the default gene-set and metabolite categories, you can manually include your own relationships for enrichment in AltAnalyze.
These analyses are available through the default AltAnalyze workflows as well as by itself as an option from the Additional Analyses menu.
Preparing Your ID List
If you are not using pre-built criterion lists from AltAnalyze (see the folder GO-Elite/regulated), you must create these files. This file should be created as a tab-delimited text file with the IDs of interest (column 1), the GO-Elite system-code (column 2) and an optional log2 fold change (column 3). If not fold change data is present, the nodes in any visualized pathways or networks will be colored yellow, otherwise, positive values will be colored red and the negative blue as shown above. The system code is a two letter code which indicates the ID system being applied. These codes are listed here. AltAnalyze can try to guess the system code if not present but this often does not work. Once finished, your file should look like this example.
| Source Identifier (REQUIRED) | SystemCode (RECOMMENDED) | Fold (OPTIONAL) |
|---|---|---|
| j05479_s_at | X | 3.2 |
| Msa.33069.0_s_at | X | -2.1 |
Performing An Enrichment Analysis
From the AltAnalyze main menu, select your species and platform. Instead of indicating the platform you can also select "Other ID" as the data type and the gene system you are analyzing as the "platform ID". Select Continue and then Additional Analyses. From here, select Pathway Analysis to see the options heatmap visualization.
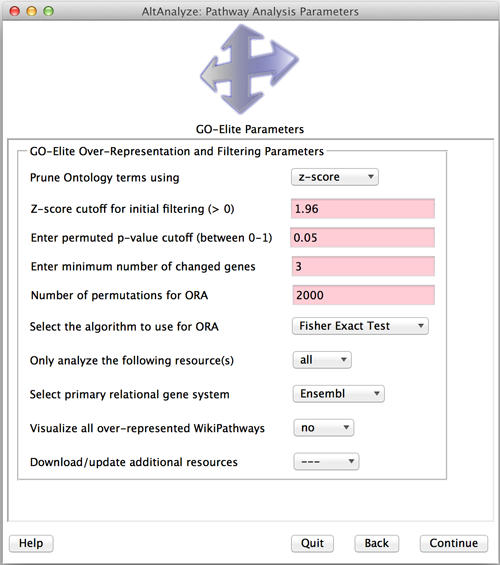
To perform an enrichment analysis:
- After creating your input file or files, save these to their own folder on your computer. If you have run an AltAnalyze workflow on unprocessed data, these files can be found in the folder GO-Elite or your input dataset files.
- (OPTIONAL) Save a denominator file(s) in a separate directory. Multiple denominators can be used with different prefixes (e.g., "set1." "set2."), where your input files also have these same prefixes.
- Select an output folder to save the results.
- Select "Continue"
- Next, you will be presented with the GO-Elite options menu. From here, you can update annotation resources or select individual resources for analysis. You can also optionally have the program generate colored WikiPathway images from your data and change the statistical methods and cutoffs used for enrichment.
- Select "Continue" to run the analysis. When finished, you will be presented with a summary of the last biological category analyzed.
Navigating the Results
There are several main result file types output by GO-Elite in the folder GO-Elite_results. These include:
- Enriched Gene-Sets and Ontology Pruned Categories with Gene IDs (pruned-results_z-score_elite.txt)
- A detailed gene ID file containing all enriched terms (pruned-gene-associations.txt)
- All original gene to pathway associations (CompleteResults/ORA)
- Overlapping statistics for different input files across different biological categories (overlapping-results_z-score_elite.txt)
- Clustered heatmaps comparing enrichment scores for all input ID lists (Heatmaps/DataPlots)
- Gene colored networks of enriched biological categories illustrating the redundancy of different genes (networks).
- Colored WikiPathways (WikiPathways).
Figure 1
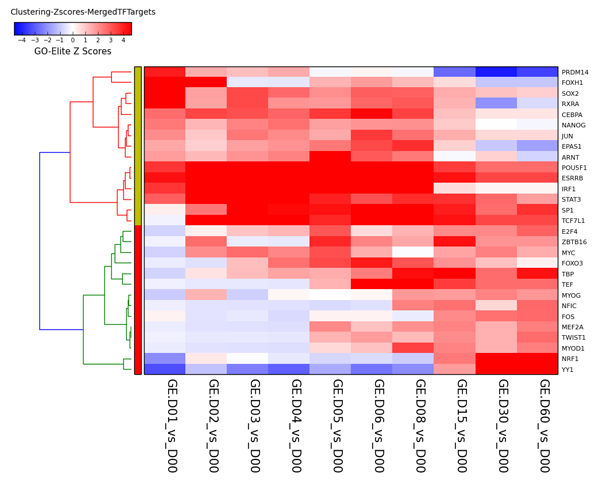
Example Result of Transcription-Factor Target Heatmap Output
Figure 2
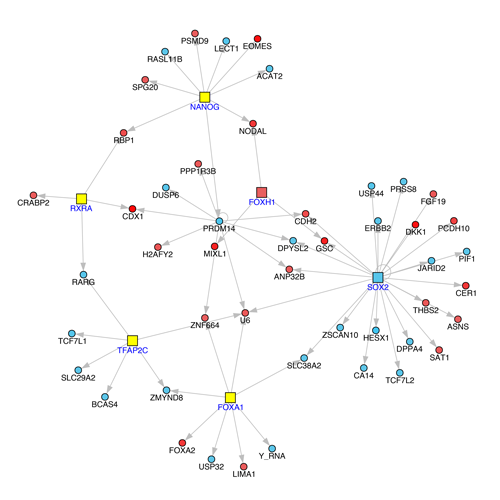
Example Result of Transcription-Factor Target Network Output
More details on each of these can be found in the GO-Elite online manual here.
GO-Elite is a software tool designed to identify a minimal non-redundant set of Gene Ontology (GO) biological terms or pathways to describe a particular set of genes. This tool calculates over-representation statistics,
Additional Options for Running GO-Elite
GO-Elite is also available as a stand-alone tool with additional options for customization, including addition of unsupported species.
GO-Elite tutorials can be found here.