Where to Save Input Expression Files?
AltAnalyze accepts multiple file formats for RNA-Seq data. In addition, this software can process specific types of microarray, proteomics or metabolomics data. The compatible file types for each include: * RNA-Seq * FASTQ files (paired-end or single-end) * BAM files (STAR, TopHat or equivalent) * 10x Genomics matrix (.mtx) files * Gene expression text files (tab-delimited .txt file) * Junction expression files (STAR, TopHat, TCGA, AltAnalyze) * Microarray * Affymetrix CEL files (various supported platforms) * Agilent (Feature Extraction files) * Gene expression text files (tab-delimited .txt file) * Proteomics/metabolomics * tab-delimited .txt file with standard molecular identifiers
When processing sample-level files in AltAnalyze, the user needs to store all of those files (e.g., FASTQ files, BAM, CEL, BED) in one directory. For FASTQ files, all gene expression and splicing analyses will be obtained from only these files. This directory can be placed anywhere on your computer and will be later selected in AltAnalyze. Example files are available for Affymetrix Exon Array Data and BED and TAB Files. Extract any downloaded TAR and/or GZIP compressed files prior to analysis.
For Affymetrix array analyses, if the user has already run normalization on their CEL files outside of AltAnalyze or have downloaded already analyzed expression data from another source, you can save the expression and DABG p-value file (optional) anywhere on your computer. These files should be tab delimited text files that only consist of probe sets, expression values and headers for each column. Example files can be downloaded here.
If beginning with a tabular expression file, simply save this file to an accessible file location and create an appropriate output directory, prior to starting AltAnalyze. If using the command-line for analysis, you will need to create you groups and comps files prior to calling AltAnalyze as described (see Groups and Comparisons and Creating Groups and Comparisons).
Running AltAnalyze from the Graphical User Interface
Windows and Mac Directions
Once you have saved your input files or normalized expression value files to a single directory on your computer, open the AltAnalyze application folder and double-click on the executable file named AltAnalyze.exe (Windows) or AltAnalyze (Mac OSX). On Mac OSX, if no application opens, click the alias icon for AltAnalyze which will bypass most machine-specific issues. These binary versions come complete will all necessary dependencies. If you installed AltAnalyze on the command-line using PyPi (pip install altanalyze), you can type altanalyze or AltAnalyze to invoke the GUI. This will open a set of user interface windows where you will be presented with a series of program options (see following sections). Note: These compiled versions of AltAnalyze can also be run via a command-line to run remotely or as headless processes (see Running AltAnalyze from the Command-Line). Once the analysis is complete, you can open the other application AltAnalyzeViewer to easily browse your results, rather than navigate the result files stored on your computer (see the ViewerManual for details or Interactive Results Viewer.
Ubuntu/Linux and Source Code Installation
On Linux and Ubuntu systems install AltAnalyze and all necessary dependencies using command-line using pip install altanalyze. To invoke the AltAnalyze GUI type altanalyze and return on the command-line. AltAnalyze can also be run from source-code (python AltAnalyze.py) but requires numerous dependencies (the program will notify you which dependencies are missing when run).
AltAnalyze Graphical Interface Options
There are many options in AltAnalyze, which allow the user to customize their output, the types of analyses they run and the stringency of those analyses. The following sections show the sequential steps involved in running and navigating AltAnalyze. For detailed descriptions of each option, see AltAnalyze Analysis Options. Interactive tutorials for different analyses are provided from the AltAnalyze website. Please note: if you will be using AltAnalyze on a machine that does NOT have internet access, follow instructions 1-5 below on an online machine and then copy the AltAnalyze program directory to an offline machine.
-
Introduction window - Upon opening AltAnalyze, the user is presented with the AltAnalyze splash screen and additional information. To directly open the AltAnalyze download page, follow the hyperlink under “About AltAnalyze”, otherwise select “Begin Analysis”.
Figure 2.1
-
Species database installation - The first time AltAnalyze is used, the user will be prompted to download one or more species database (requires internet connection). Independent of the data source (e.g., RNASeq or array type) you are analyzing, select a species and continue. The user can select from different versions of Ensembl. If your species is not present, select the button Add New Species. Selecting the option Download/update all gene-set analysis databases will additionally download GO-Elite annotation databases needed for performing a wide-array of biological enrichment analyses (pathways, ontologies, TF-targets, miR-targets, cellular biomarkers) and network visualization analyses.
Figure 2.2

-
Select species and platform - Next, the user must select a species, array vendor or data type and platform for analysis (Figure 2.3). Array vendors include Affymetrix, Illumina, Agilent and Codelink. The data type, Other ID can be selected if loading non-normalized or normalized values from a different data source (select ID type under Select Platform). This applies also to RNA-Seq normalized gene values from a different workflow, such Cufflinks, eXpress or RSEM. In these cases, match the input ID type (e.g., Symbol, Ensembl) under Select Platform. If multiple database versions have been downloaded, the user will also be able to select a version pull-down menu. After selecting these options click Continue.
Figure 2.3
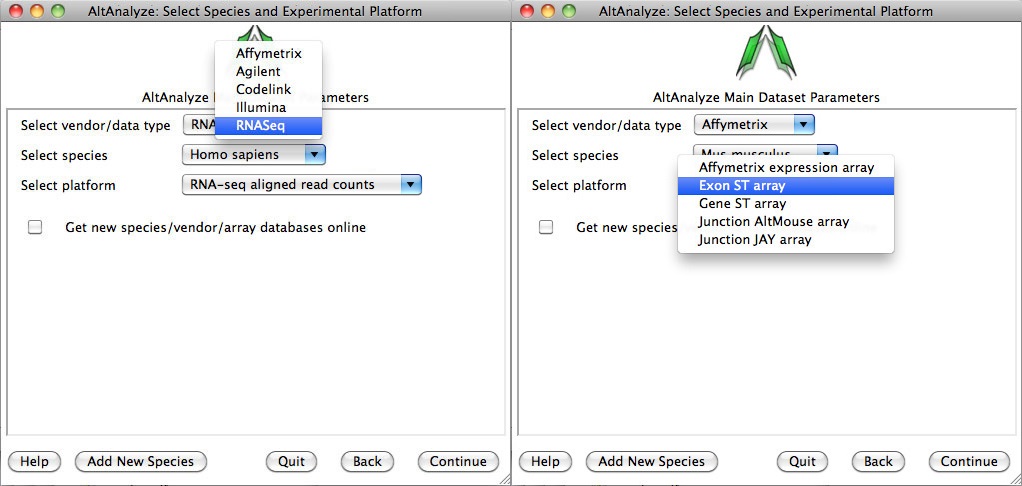
Select a species and vendor or data type for analysis: (Left) Options for selecting currently supported species, array vendors or data types (RNASeq). Select the check box to download the latest AltAnalyze gene and exon databases.
-
Select analysis option - In this window, the user must select the type data being analyzed. There are four main types of data: 1) FASTQ, BAM, BED, CEL or Feature Extraction files, 2) Expression files (normalized or read counts), 3) AltAnalyze filtered files and 4) results from third-party applications (Annotate External Results). Alternatively, the user can open the Interactive Results Viewer or perform Additional Analyses. Processing of CEL files will produce the two file types (expression and DABG), while processing of BED files will produce a file of exon and/or junction counts. Processing of Expression files, allows the user to select tab-delimited text files where the data has already been processed (e.g. RMA or read counts), which will also produce AltAnalyze filtered files. AltAnalyze filtered files are written for any splicing array analysis (not for gene expression only arrays). These later files allow the user to directly perform splicing analyses, without performing the previous steps. The AltAnalyze filtered files are stored to the folder “AltExpression” under the appropriate array and species directories in the user output folder. Since CEL file normalization and array filtering and summarization can take a considerable amount of time (depending on the number of arrays), if re-performing an alternative exon analysis with different parameters, it is recommended that the user select the Process Expression file or Process AltAnalyze filtered, depending on which options the user wants to change. Users can also import lists of regulated probe sets with statistics obtained from a third-party application (e.g., JETTA) other than AltAnalyze using the Annotate External Results option. In addition, expression clustering, pathway visualization, pathway enrichment and lineage classification can be independently run on a user expression file using the option Additional Analyses.
Figure 2.4
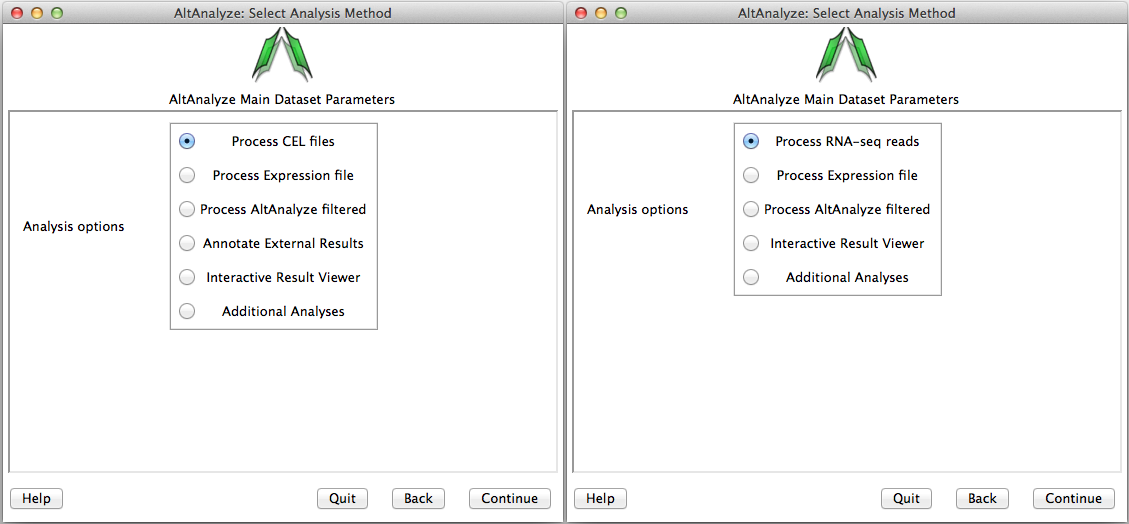
Select the analysis type: Options available for the select species and array or data type. AltAnalyze filtered is only available for alternative exon analyses.
-
Processing CEL, feature extraction or exon/junction files - If you selected the first option from "Main Dataset Parameters", you will be presented with a new window for selecting the location of your CEL/FE/BED/TAB files and desired output directory. Clicking the "select folder" icon will allow you to browse your hard-drive to select the folder with these data files. You can double-check the correct directory is selected by looking at the adjacent text display. For Agilent Feature Extraction files, you will be presented with the option of selecting which channels or channel ratios to extract data from. For Affymetrix CEL files, this window will be followed by an indicator window that will automatically download the library and annotation files for that array. If the array type is unrecognized and you do not already have Affymetrix library files for your array (e.g. PGF or CDF), you will need to download these files from the Affymetrix website. To do so, select the link at the bottom left side of this window named "Download Library Files". Select the array type being analyzed from the web page and select the appropriate library files to download and extract to your computer (requires an Affymetrix username and password) (Figure 2.5 Bottom). For RNA-Seq data, the user selects the folder containing their exon and junction input files in either BED (e.g., TopHat) or TAB (BioScope) files. If the user is analyzing junction alignment results but does not have exon-level results, the user can build an annotation file for the program BEDTools, to derive these results from an available BAM file (e.g., produced by TopHat). To do this, select the option "Build exon coordinate bed to file obtain BAM file exon counts". Instead of running the full AltAnalyze pipeline, AltAnalyze will immediately produce the exon annotation file for BEDTools to the BAMtoBED folder in the user output directory. See Building a Dataset Exon Database for BEDTools for additional details.
Figure 2.5
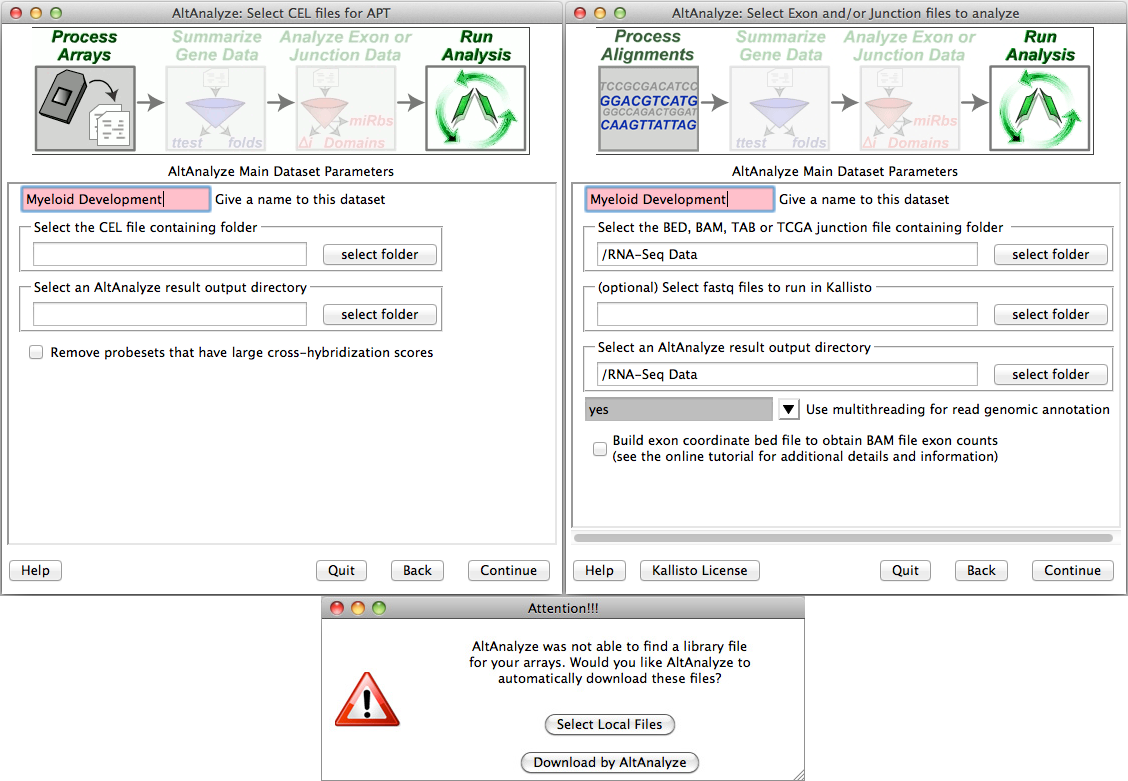
Select folder and file locations: (Top) Pre-built BED or TAB files are currently required to run AltAnalyze on RNA-Seq data. To properly run RMA using the APT software (included with AltAnalyze), the user must select a valid folder containing CEL files. (Bottom) The first time you analyze a certain type of array you will be prompted to download a library file(s) for that array. For some arrays, you will be prompted for AltAnalyze to download these plus annotation files for you. Otherwise, you will be prompted by the program for such files.
-
Summarizing gene data and filtering for expression - After obtaining summary read counts or normalized CEL expression values, a number of options are available for summarizing gene level expression data, filtering out RNA-Seq reads and probe sets prior to alternative exon analysis and performing additional automated analyses (Figure 2.6). Selection of a comparison group test statistic, allows the user to calculate a p-value for gene expression and splicing analyses based on different tests (e.g., paired versus unpaired t-test). Batch-effect correction can be optionally performed with the combat library. For applicable platforms, the option to perform quantile normalization is also provided here. For Affymetrix splicing arrays, AltAnalyze calculates a “gene-expression” value based on the mean expression of all “core” (Affymetrix core probe sets and those aligning to known transcript exons) or “constitutive” (probe sets aligning to those exon regions most common among all transcripts) probe sets that have a mean DABG p-value less than and a mean expression value greater the user indicated thresholds for each gene. The same methods are used for RNA-Seq exon or junction counts, using the same Ensembl and UCSC combined constitutive evidence. Rather than observed counts, RPKM normalized counts are selected by default, with counts as an alternative options (see Algorithm Descriptions). These values are used to report predicted gene expression changes (independent of alternative splicing) for all user-defined comparisons (see following section). In addition, fold changes and ttest p-values are calculated for each of these group comparisons. These statistics along with several types of gene annotations exported to a file in the folder “ExpressionOutput” in the user-defined results directory. Along with this tab-delimited text file, a similar file with those values most appropriate for import into the pathway analysis program GenMAPP will also be produced (Figure 5.1). For splicing analyses, RNA-Seq reads or probe sets with user defined splicing cutoffs (expression and DABG p-values) will be retained for further analysis (see ExpressionBuilder Module). Other analyses, such as QC, PCA, hierarchical clustering (significant genes and outliers), prediction of which cell lineages are detected and pathway analysis are also automatically run using these options. If the user selects no for any these, they can be run again later using the Additional Analyses option from the Select Analysis Method menu.
Figure 2.6
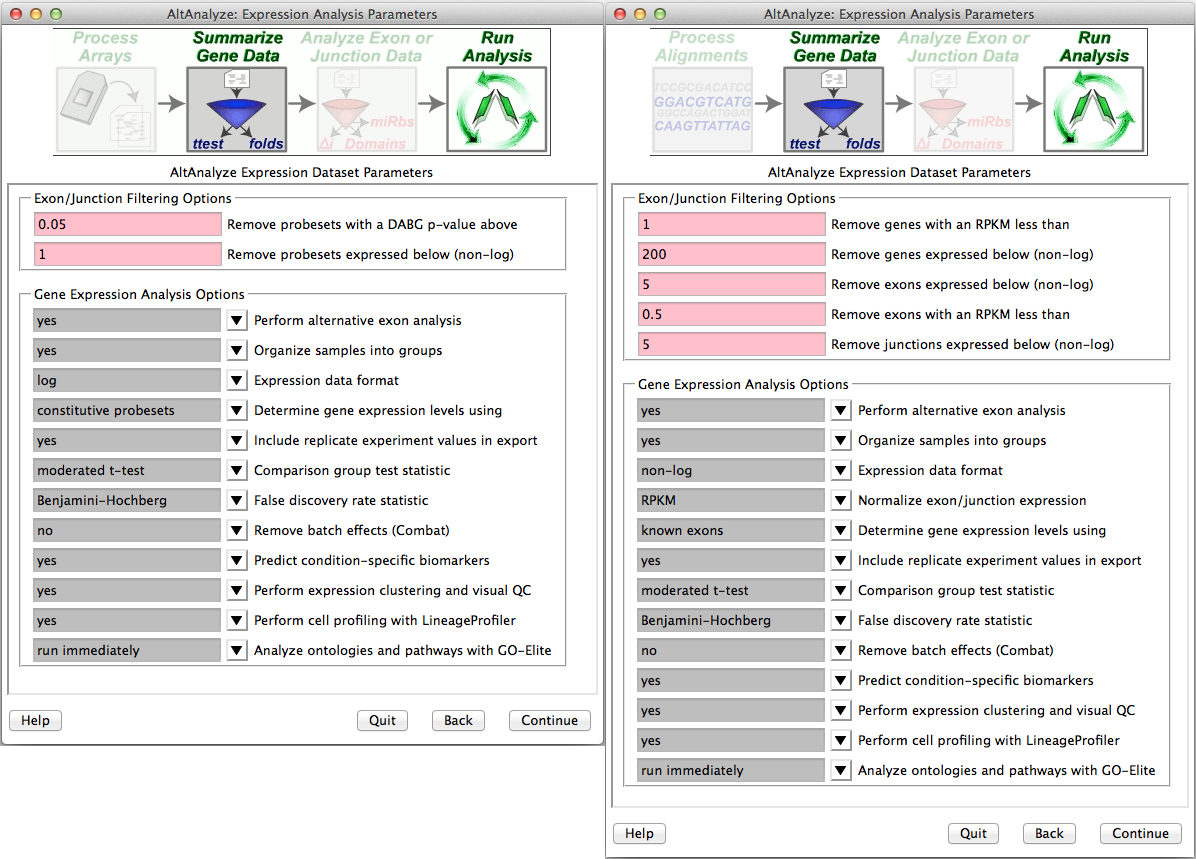
Select summarization and filtering options: Users are presented with options for filtering RNA-Seq reads or probe sets for alternative exon analyses (DAGB and mean group expression) and options for how to derive gene expression values.
-
Select alternative exon analysis parameters: - If using a junction (RNA-Seq or junction array) or exon-sensitive array (e.g., Human Affymetrix Exon 1.0 or Gene 1.0 ST), the user will be presented with specific options for that platform (Figure 2.7). These options include alternative exon analysis methods, statistical thresholds, and options for additional analyses (e.g., MiDAS), however, the default options are typically recommended. Users can also choose whether to analyze biological groups as pairwise group comparisons, comparison of all groups to each other or both. These include combining values for exon-inclusion junctions and restricting an analysis to a conservative set of Affymetrix probe sets (e.g., core) and changing the threshold of splicing statistics. Note: that AltAnalyze’s core includes any probe set associated with a known exon. When complete, the user can select “Continue” in AltAnalyze to incorporate these statistics into the analysis.
Figure 2.7
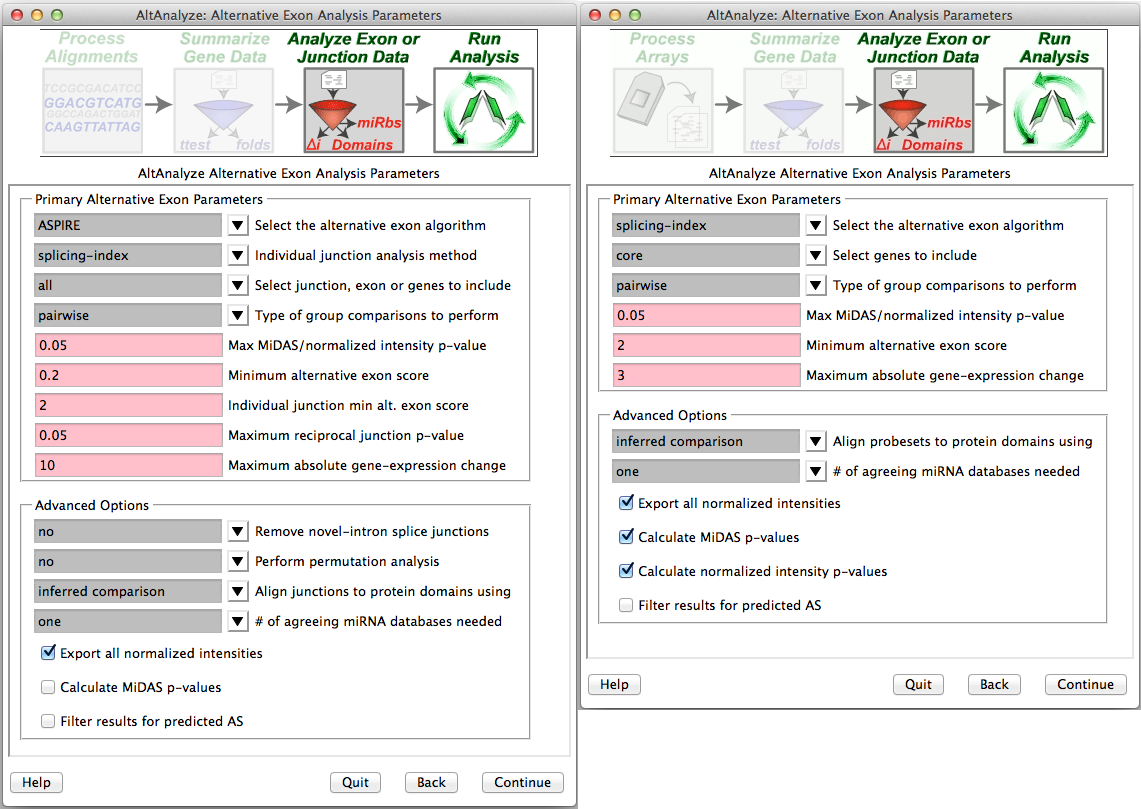
Select alternative exon analysis options: Splicing analysis options for (Left) RNA-Seq or junction arrays and (Right) exon arrays (e.g., Exon 1.0 and gene 1.0).
-
Assigning groups to samples - When analyzing a dataset for the first time, the user will need to establish which samples correspond to which groups. Type in the name of the group adjacent to each sample name from in your dataset (Figure 2.8 Left). When selecting batch-effect correction (combat), an additional menu similar to the group annotation will appear afterwards asking the user to enter the batch effect for each one. For single-cell datasets or other datasets where you wish to predict de novo groups, select the Run de novo cluster prediction (ICGS) to discover groups option to discover clustered sample groups for further analysis in AltAnalyze (see Step #11 below and Algorithm Descriptions for details.)
Figure 2.8
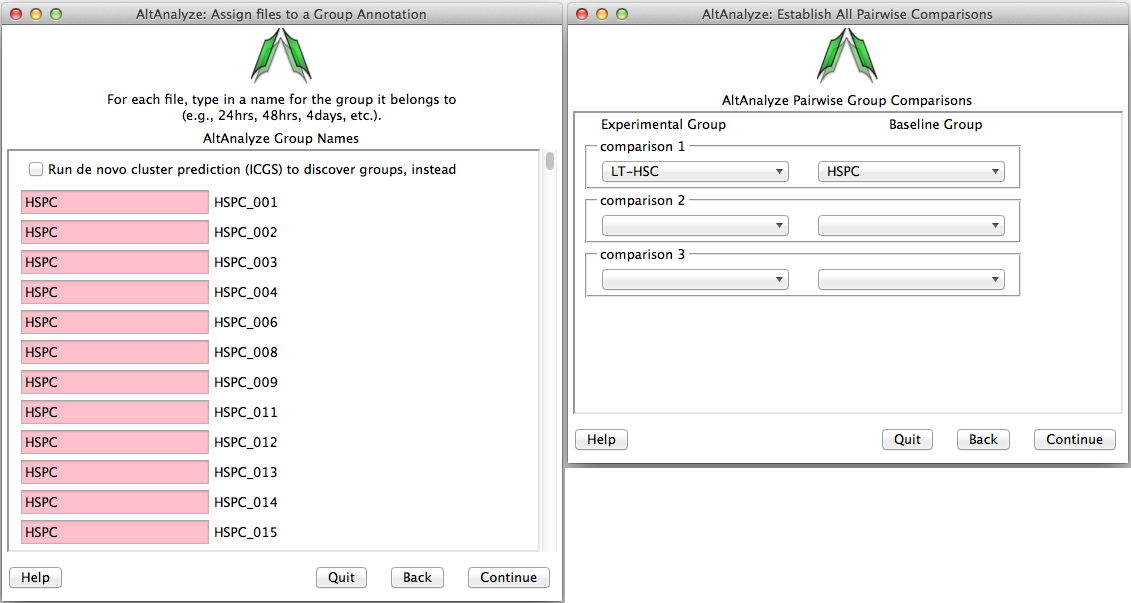
Establish groups and comparisons: (Left) Enter a name for each group for all samples. Optionally, select “Predict Groups from Unknown Sample Types” to predict groups. (Right) Enter all group comparisons for any possible pairwise group comparisons (in this case there is only one). These relationships can be created in advance in a spreadsheet program for command-line analysis in AltAnalyze or for large sample datasets. For more details see Creating Groups and Comparisons.
-
Establishing comparisons between groups - Once sample-to-group relationships are added, the user can list which comparisons they wish to be performed (Figure 2.8 Right). For splicing and non-splicing arrays, folds and p-values will be calculated for each comparison for the gene expression summary file. For RNA-Seq read or splicing arrays, each comparison will be run in AltAnalyze to identify alternative exons. Thus, the more pairwise comparisons the longer the analysis. If the user designates to compare “all groups” and not designate a pairwise comparison, this window will not be displayed.
-
AltAnalyze status window - While the AltAnalyze program is running, several intermediate results files will be created, including probe set or RNA-Seq read, gene and dataset level summaries (see AltAnalyze Analysis Options). The results window (Figure 2.9) will indicate the progress of each analysis as it is running. When finished, AltAnalyze will prompt the user that the analysis is finished and a new “Continue” button will appear. A summary of results appears containing a basic summary of results from the analysis. This window contains buttons that will open the folder containing the results and suggestions for downstream interpretation and analysis. Selecting the button “Start DomainGraph in Cytoscape” will allow the user to directly open a bundled version of Cytoscape and DomainGraph (see Analysis of AltAnalyze Results DomainGraph). In addition to viewing the program report, this information is written to a time-stamped log text file in the user-defined output directory.
Figure 2.9
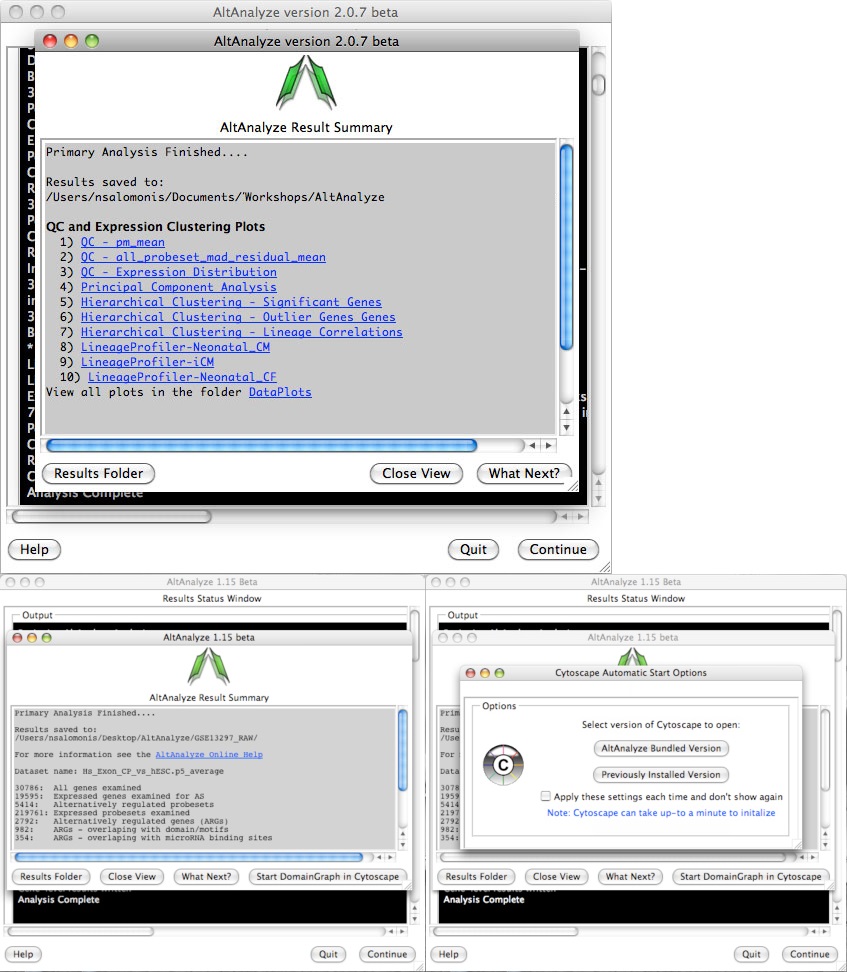
AltAnalyze status: The AltAnalyze status window will appear once all user options are defined. Analysis run-time will depend on the number of samples, comparisons and array type. (Top) Gene expression analysis results are shown for Affymetrix array data (alternative exon analysis omitted). Links to data plots (PNG files) are shown for the default selected QC, clustering and lineage analyses. (Bottom) Summary of alternative exon results is shown for an exon or junction-sensitive platform. By selecting the option Start DomainGraph in Cytoscape, users can immediately proceed to results visualization in DomainGraph at the level of transcripts, proteins, domains, exons and microRNA binding sites.
-
Analyze ontologies and pathways with GO-Elite - If the user selects this option during the analysis or following, they will be presented with a number of options for filtering their expression data to identify significant regulated genes, perform pathway, ontology or gene-set over-representation analyses and filter/prune the subsequent results. Selecting to visualize WikiPathways may significant time to the analysis. Regulated alternative exons will also be analyzed using GO-Elite. A similar summary results window as above will also appear with the GO-Elite WikiPathways and Gene Ontology results (Figure 2.10). For additional information, see http://genmapp.org/go_elite.
Figure 2.10
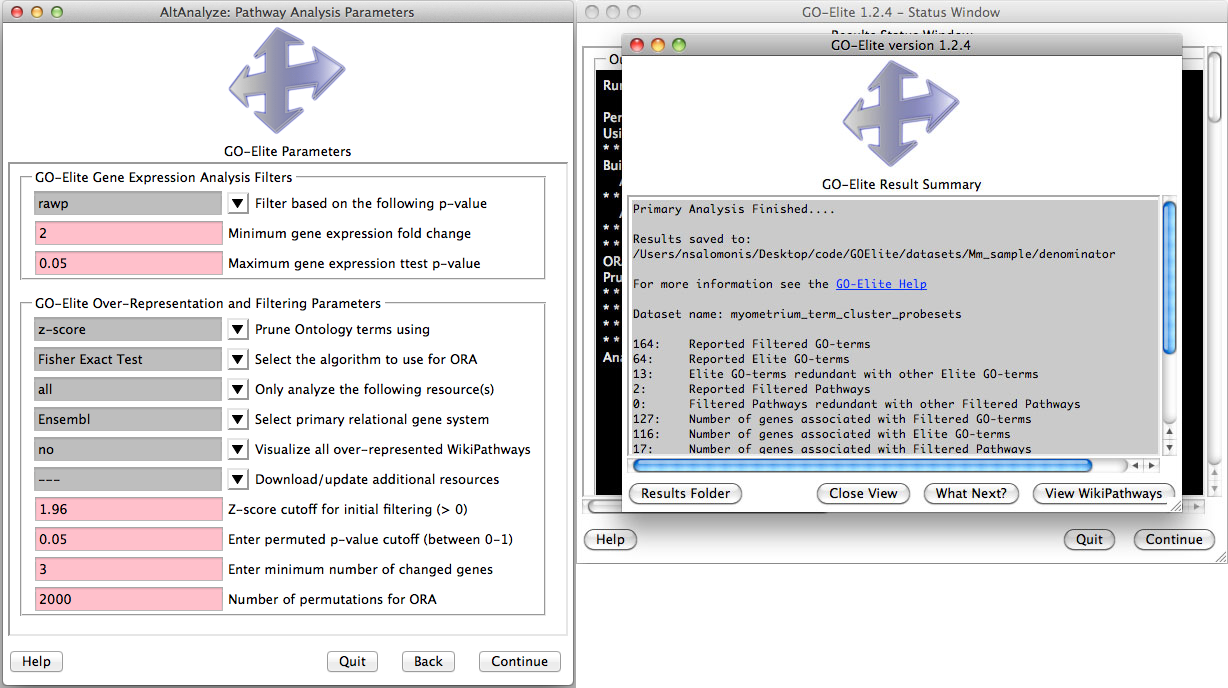
Perform pathway over-representation analysis: (Left) Options to analyze both differentially expressed and alternative expressed genes from AltAnalyze summary statistics. Options include how stringent the gene expression statistics are, whether or not to visualize pathways and methods for redundancy filtering between Gene Ontology (GO) terms from the program GO-Elite. Additional ontologies (Disease, Phenotype), pathways (KEGG, PathwayCommons) and gene-sets (e.g., BioMarkers, transcription factor targets) can be updated and analyzed. (Right) Results are reported for GO and WikiPathways are reported when the user runs GO-Elite after the primary analysis.
-
Predict groups from unknown populations (ICGS) - When a priori sample groups are unknown, such as with Single-Cell RNA-Seq analyses, it is recommended that the user discover sample groups clustered based on highly distinct gene or alternative isoform expression patterns. This can be accomplished by selecting the Run de novo cluster prediction (ICGS) to discover groups option from the Group selection menu. This menu implements a robust algorithm for iteratively filtering, correlating and clustering the data to find coherent gene expression patterns that can inform which sample groups are present. Iterative Clustering and Guide-gene Selection (ICGS) identifies the predominant correlated expression patterns from a given gene or splicing dataset to identify predominant, rare and transitional cell or tissue states. The resulting menu (Figure 2.11), will present the user with options for filtering their dataset (RNA-Seq or other input datasets), based on the maximal non-log expression for each row (Gene TPM or RPKM filer cutoff), number of associated reads for each gene (if applicable, otherwise set equal to the above), minimum required fold change difference between the minimum and maximum expressed samples for each gene and associated number of samples for this comparison, correlation threshold between genes for identification of coherent gene set clusters, which features to evaluate (gene, alternative exons, or both) and which gene sets to optionally build off. Although designed for RNA-Seq, any datasets can be analyzed with these menu options. The results will be presented in the form of clustered heatmaps, from which you can select from different options. Each heatmap will have somewhat distinct sample clusters from which you can select to perform the conventional AltAnalyze comparison analysis workflows, using the parameters established in the prior menu options. As results can vary based on the clustering algorithm used, we recommend that you have R installed on your computer (not required), to use the HOPACH clustering algorithm which provides improved results. For Windows operating systems, R is now included with the binary version of AltAnalyze. Additional information on ICGS and video tutorials can be found here.
Figure 2.11
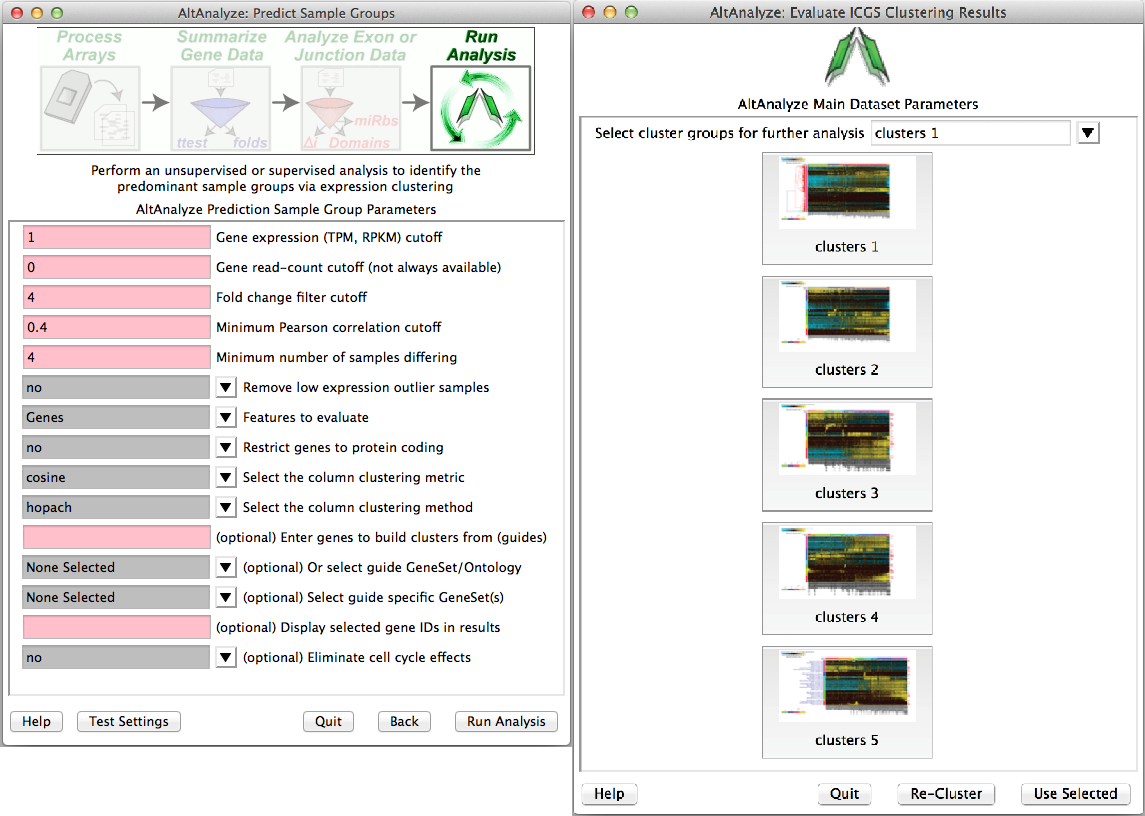
Iterative clustering and guide-gene selection (ICGS): (Left) Available options for unsupervised and supervised identification of differentially regulated gene or splicing sets and associated sample populations from the ICGS menu. Analysis options include the selection of specific pathways from which to correlate to all other genes to build coherent gene clusters (supervised analysis), removal of cell-cycle effects and initial filtering and correlation parameters. (Right) Resulting hierarchically clustered results from which to select from for further downstream comparison analyses in AltAnalyze. Selection of each icon will preview the entire cluster (specific genes can be identified from the resulting files saved in the ICGS directory). Cluster options can be selected from the top pull-down menu for further comparison analyses.
AltAnalyze Viewer
AltAnalyze is now distributed with an integrated application called the AltAnalyze Viewer which allows users to immediately and interactively navigate the results from an AltAnalyze workflow analysis (see above). This viewer allows the user to navigate all heatmap images, networks, colored pathways, quality control plots and result tables. In addition, various interactive plots can be called from this viewer using built-in AltAnalyze functions, including heatmaps, PCA, SashimiPlots, Domain and exon expression plots (Figure 2.12). Tables themselves can be interactively searched for genes and expression data plotted. An example use of the viewer is shown here.
Figure 2.12
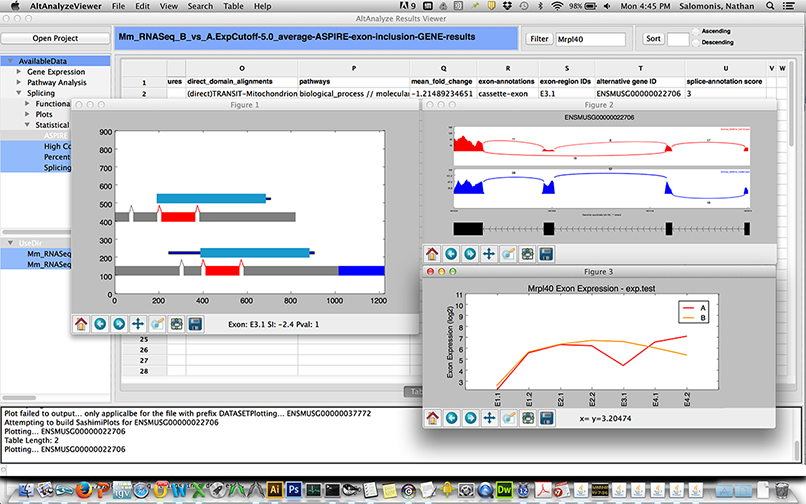
AltExon Visualization in the Results Viewer application: The AltAnalyze exon viewer can be initiated by selecting the executable from the AltAnalyze program folder or through the AltAnalyze main menu. Selection of various interactive splicing plots in the AltAnalyze Viewer from an ASPIRE splicing result table view (right click option).
Additional Analysis Options
Many of the analysis tools present in AltAnalyze can be run independent of the above described workflows on user input text files. The input text files are either a table of log2 expression values, fold-changes or identifiers for over-representation analysis in GO-Elite. The options are available from the menu Additional Analyses from the menu Select Analysis Methods (Figure 2.13) as well as from the command-line. In addition to the below overviews, more information on these methods and available options can be found in Overview of Analysis Results and Additional à la carte Analyses.
Figure 2.13
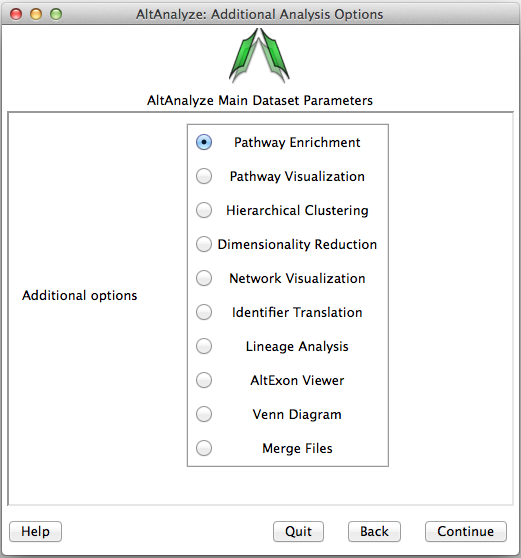
Additional analysis options: Analyses that can be run on any properly formatted user data. This includes pathway over-representation, visualization, hierarchical clustering, principal component, lineage analysis, network visualization, alternative exon graphs, Venn diagrams, identifier translation and file merging options.
-
Pathway Enrichment - Performs GO-Elite analysis as described in the previous section on any existing directory of input and denominator identifiers. This method runs GO-Elite independent of other AltAnalyze functions. In addition to saving lists of enriched biological categories, this tool produced hierarchically clustered heatmaps of enriched terms between input ID lists along with network graphs displaying interactions between genes and enriched pathways, ontology terms or gene sets. When the pathway visualization option is also selected, all enriched WikiPathways will also be exported as colored PDF and PNG images. If selecting already produced GO-Elite inputs produced by AltAnalyze, see the folder GO-Elite/input in the AltAnalyze results folder.
-
Pathway Visualization - Using the same GO-Elite input files, the user can select any current WikiPathway and visualize log2 fold changes on the selected pathway through the AltAnalyze user interface. The input file must have three columns (ID, SystemCode, FoldChange). Images will be saved as PNG and PDF files to the same directory as the input file (Figure 2.14). When running from source-code, ensure that the python packages lxml and requests are installed (see Installation).
Figure 2.14
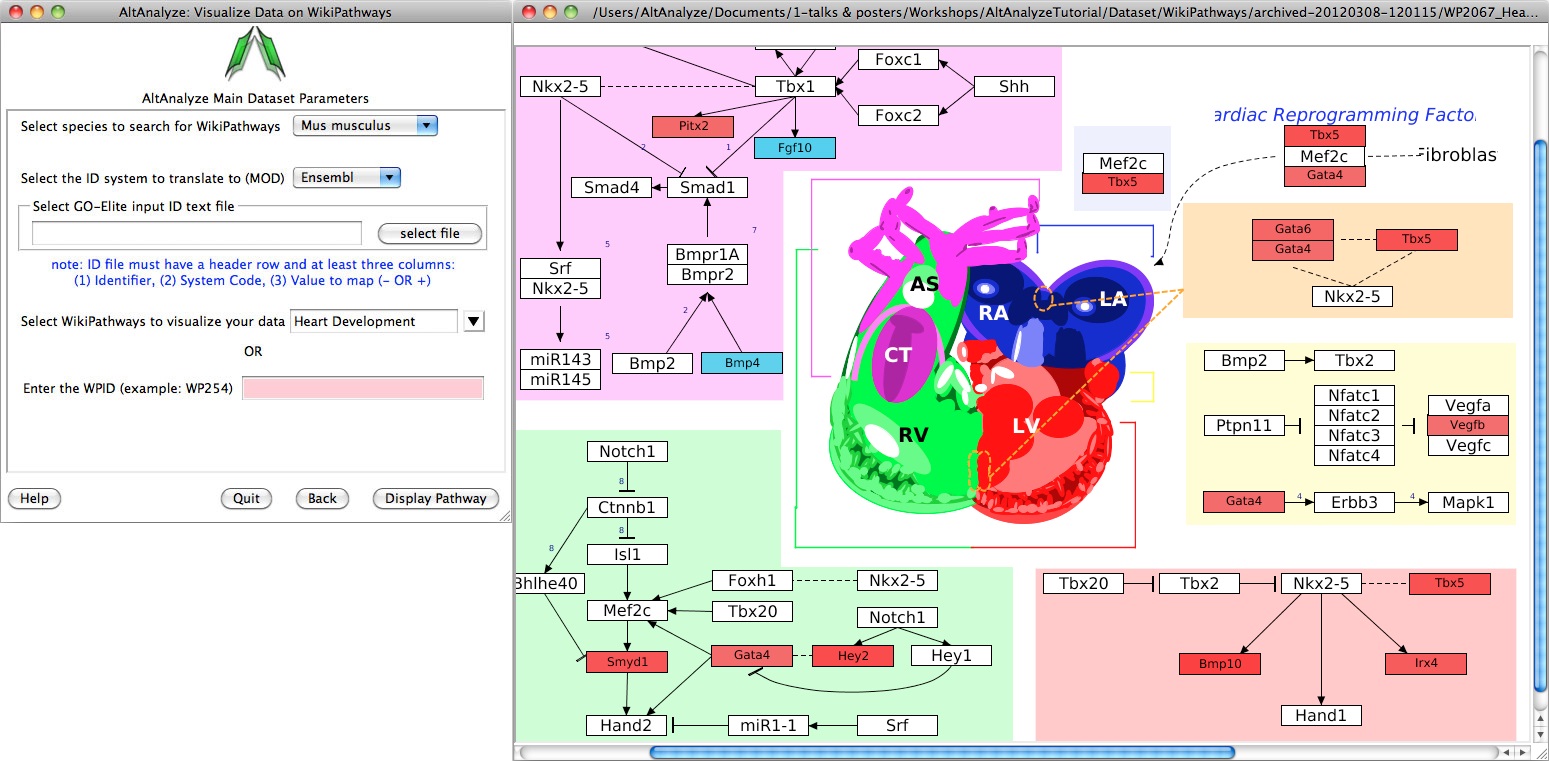
Visualize WikiPathways in AltAnalyze: Users can select pathways to visualize their GO-Elite input files upon for any WikiPathway. (Left) The selection window opened from the Additional Analyses menu and (Right) an example visualized pathway is shown. Default red = positive values, blue = negative values.
-
Hierarchical Clustering - This interface will output a clustered heat map of rows and columns for any user supplied input text file (Figure 2.15). This file must have column names (e.g., samples) and row names (e.g., probesets), with the remaining data as values. The user can choose the clustering algorithm or metrics to use, whether to cluster rows or columns and what colors to use. This algorithm is automatically run when using the default AltAnalyze workflows on two gene-sets: 1) all significantly differentially expressed genes and 2) outlier regulated genes. These files are available in the folder ExpressionOutput/Clustering. Significantly differentially expressed genes in these sets are defined as > 2 fold (up or down) regulated and comparison statistic p < 0.05 (any comparison), unless the options are changed in the GO-Elite interface. Outlier genes are those with > 2 fold (up or down) regulated in any sample relative to the mean expression of all samples for that gene and not in the significantly differentially expressed list. Many additional advanced options, including filtering by pathways, ontology terms and other gene sets and single-cell discovery analysis options are described in Additional à la carte Analyses. Resulting clusters are interactive, allowing for viewed genes to be explored in online databases, pathways to be evaluated for associated genes and connections and deeper visualization in TreeView by selection of the TreeView viewer option in the lower left hand corner of the heatmap. Additionally, visualization of pre-assigned sample groups can be viewed by adding the group prefix prior to the sample name as a colon separated annotation (e.g., group_name:sample_name) or analyzing the expression files in the directory ExpressionInput.
Figure 2.15
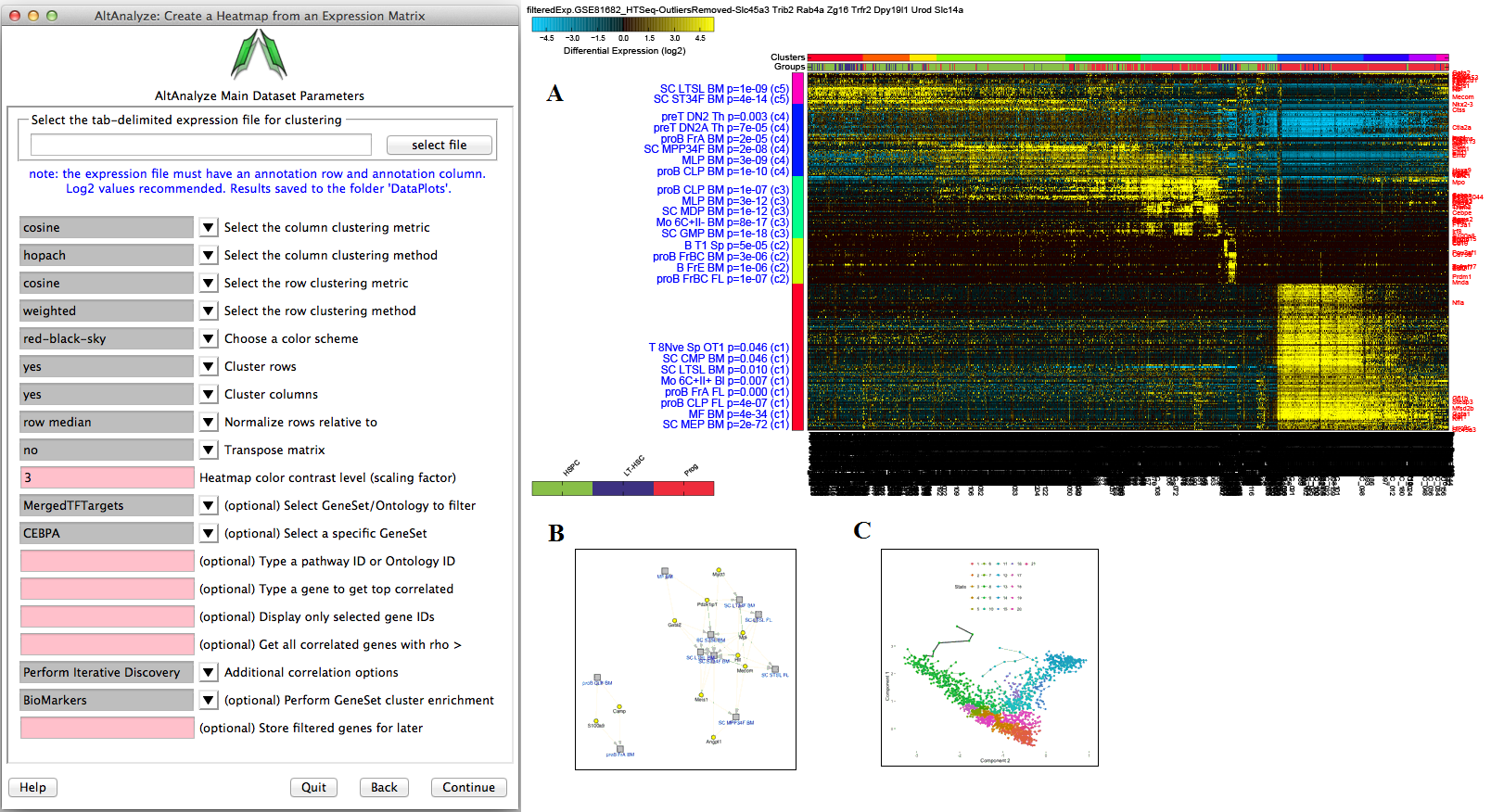
Hierarchical Cluster and Heatmap Visualization: This function can be used to identify global patterns of expression from any user input text file. (Left) The basic and advanced parameters for clustering and visualization or shown as well as (A) an example heatmap derived by clustering columns and rows from an input text file of log2 folds, associated enriched pathways on the left and genes used selected by the software to build clusters from on the right (optional). In this example, ~1,700 single-cell RNA-Seq libraries, were analyzed with ICGS in AltAnalyze[Nestorowa2016] were cluster using HOPACH clustering (requires that R is installed) and visualized with enriched ImmGen delineated marker genes (MarkerFinder determined in AltAnalyze). Guide-genes are displayed in red to the right of each cluster. Additional advanced options are described in Additional à la carte Analyses. Default red = positive values, blue = negative values. (B) Gene-to-ImmGen category network. Upon selection of an enriched term for a gene cluster (blue text - panel A), a window will appear with a list of the genes in the selected term (genes copied automatically to clipboard) followed by the opening of a png file with a displayed network showing all gene to term relationships for that gene cluster. The genes and terms shown correspond to gene cluster 5 (c5 - short term and long term stem cells). (C) Monocle analysis results for the ICGS clusters and genes shown in panel A. Monocle analysis in R will be automatically run if the user selects the option Monocle from the Additional correlations option. These results will be saved to DataPlots/Monocle in the input file directory (installation of R is required for Mac and Linux operating systems).
-
Dimensionality Reduction Analysis - Multiple options for dimensionality reduction are available in AltAnalyze. These include two and three-dimensional principal component visualization (using Z-score normalization) and t-SNE (Figure 2.16). The top 100 correlated and 100 anti-correlated genes for top 4 PCs (~800 total, some redundant) with each principal component can be stored by entering a name for the analysis options menu, for further analysis in the above hierarchical clustering tool. Additionally, specific genes can be entered into this interface to color those genes based on their relative expression within the PCA scatter plot. This analysis is useful for determining how similar samples, individual cells and biological groups are two each other in the 2D or interactive 3D space (see Additional à la carte Analyses for more details).
Figure 2.16
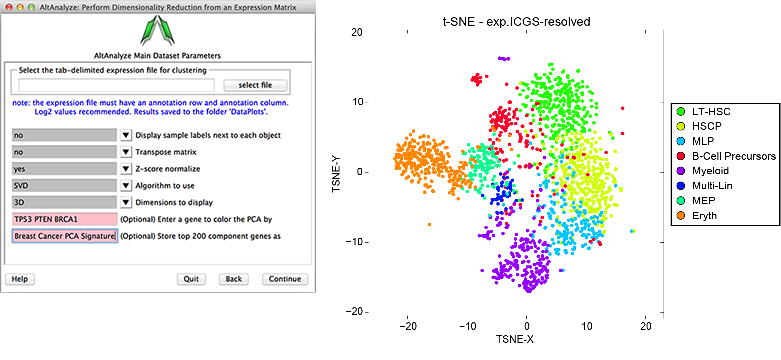
Dimensionality Reduction Analysis: This function can be used to perform either PCA, SVD or t-SNE dimensionality reduction, associated visualization and PC loading gene identification. (Left) Example parameter options are shown in a screenshot of this menu. (Right) T-SNE analysis ICGS derived cell clusters obtained from the single-cell RNA-Seq analysis of hematopoietic progenitors[Nestorowa2016].
-
Lineage Analysis - This option allows the user to identify correlations to over 70 tissues and cell types for a group of biological sample. The input file must be tab-delimited and have expression values (log2 for microarray datasets) for each array (e.g., probeset) identifier. Visualization of these results is provided for Z scores calculated from the lineage correlation coefficients upon a comprehensive Lineage WikiPathways network and as a hierarchically clustered heat map. Additional options for alternative modes of sample classification and custom reference sets are described in Additional à la carte Analyses.
Figure 2.17
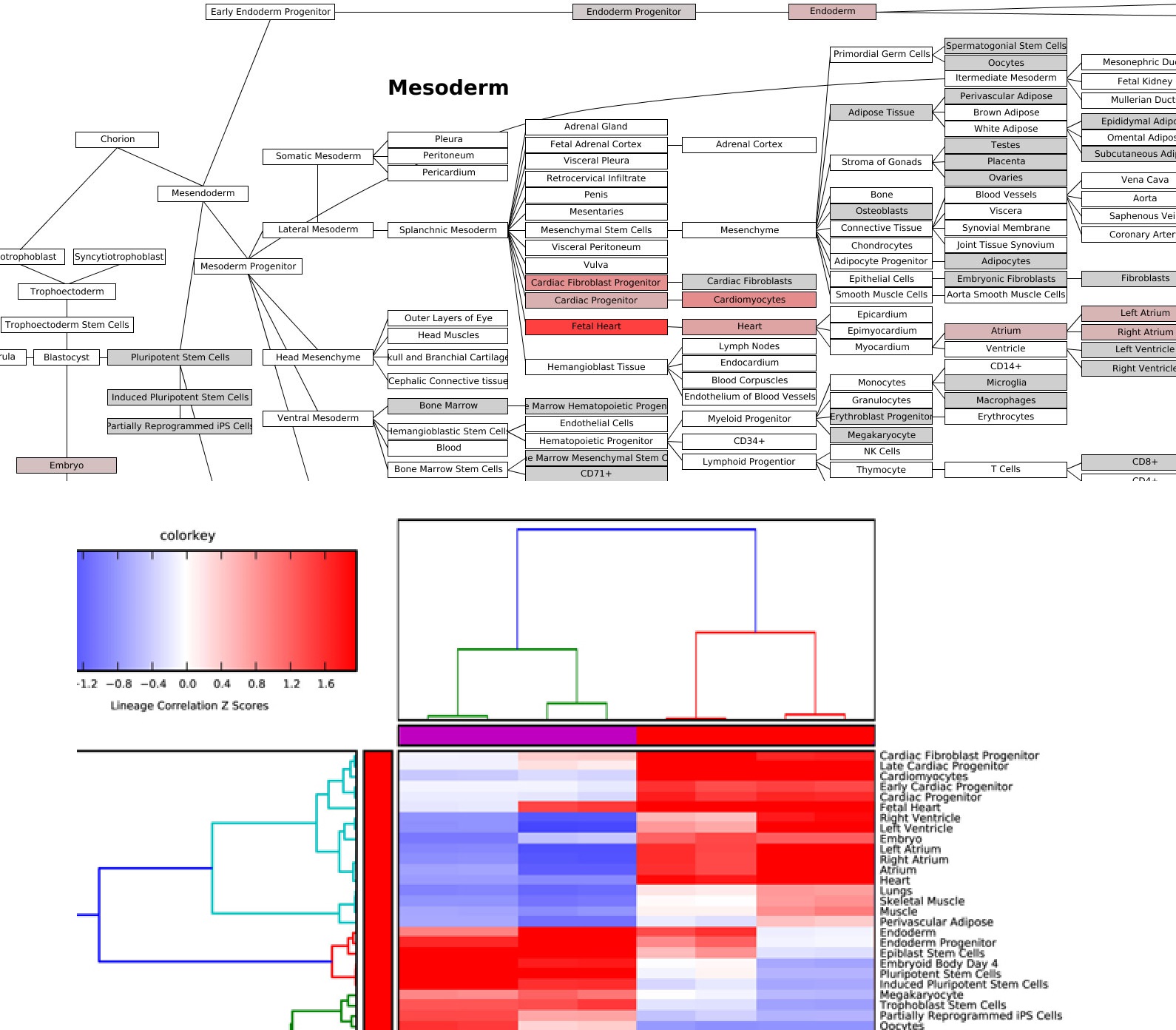
Lineage Profiler Analysis: Lineage Profiler output for RNA-Seq cardiac differentiation time-points gene expression data. Visualization of correlation-based Z scores for (Top) a single differentiation time-point along a comprehensive lineage network and (Bottom) comparison of lineage associations for all samples examined after hierarchical clustering.
-
Network analysis - This option (aka NetPerspective) allows users to build and view biological interaction networks built using input sets of genes, protein, metabolite identifiers along with data indicating the regulation of these genes. See Additional à la carte Analyses for more details.
-
Venn Diagram visualization - To identify the overlap between identifiers found in two or more files, users can select this menu options to obtain overlapping Venn Diagrams of the IDs overlapping in distinct files. Two methods are available for visualization of these diagrams: (A) Standard overlapping Venn’s and (B) ID membership weighted (see Additional à la carte Analyses for more details).
-
Alternative Exon Visualization - This method allows users to view either raw exon expression (e.g., RPKMs, probeset intensity) or gene-normalized normalized expression values (splicing-index) for all exon-regions for a given set of genes. Users must first select the AltResults folder from a given experiment. When more than two groups of samples are present in a given study, it is recommended that the user also perform the alternative exon anlaysis for all group comparisons (rather than pairwise) to simultaneously view all biological groups. When viewed in this context, distinct sample groups are displayed as different colored lines with error bars indicated by the standard error. Individually entered genes or files containing many genes can be displayed or saved to the users hard disk (exported to the folder ExonPlots). For more details, see Additional à la carte Analyses.
-
Identifier Translation - This method can be used to translate from one gene, protein or metabolite ID system to another. Simply load a file of interest and select the input ID system and output ID system. A new file will be saved to the same directory in which the input file is in with the extension name of the output ID system.
-
Merge Files - This function allows users to identify sets of IDs that overlap or that are distinct from each other from a set of distinct files. As many as four files can be selected, using the options Union or Intersection.
Running AltAnalyze from the Command-Line
In addition to using the default AltAnalyze graphical user interface (GUI), AltAnalyze can be run by command line options by calling the python source code in a terminal window or through other remote services. This option can be used to run AltAnalyze on a remote server, to batch script AltAnalyze services or avoid having to select specific options in the GUI. To do this, the user or program passes specific flags to AltAnalyze to direct it where files to analyze are, what options to use and where to save results.
Methods for Command-Line Processing
-
When installing source code, run from within the AltAnalyze program directory by calling AltAnalyze.py followed by command-line arguments.
-
When running with OS-specific binaries of AltAnalyze directly call the binary files themselves:
-
Windows OS:
AltAnalyze.exe -
Mac OS X:
AltAnalyze.app/Contents/MacOS/AltAnalyze -
Ubuntu OS:
./AltAnalyze
-
Examples and Flag description
For detailed examples, flag descriptions, default values and associated information, see Running AltAnalyze from Command Line Interface.
Downloading and installing a species-specific database (mouse)
python AltAnalyze.py --species Mm --update Official --version EnsMart62
--additional all
Analyzing RNA-Seq files – FASTQ file directory using ICGS Population Discovery
python AltAnalyze.py --runICGS yes --platform "RNASeq" --species Mm
--column_method hopach --rho 0.4 --ExpressionCutoff 1 --FoldDiff 4
--SamplesDiffering 4 --excludeCellCycle conservative --output "C:/FASTQ_Files/"
--expname "Mm_HSCs" --fastq_dir "C:/FASTQ_Files/" --runKallisto yes
Analyzing RNA-Seq files – BAM file directory using ICGS Population Discovery
python AltAnalyze.py --runICGS yes --platform "RNASeq" --species Mm
--column_method hopach --rho 0.4 --ExpressionCutoff 1 --FoldDiff 4
--SamplesDiffering 4 --excludeCellCycle conservative --output "C:/FASTQ_Files/"
--expname "Mm_HSCs" --bedDir "C:/FASTQ_Files/"
Analyzing CEL files - Affymetrix 3’ array using default options and GO-Elite
python AltAnalyze.py --species Mm --arraytype "3'array" --celdir "C:/CELFiles"
--groupdir "C:/CELFiles/groups.CancerCompendium.txt"
--compdir "C:/CELFiles/comps.CancerCompendium.txt"
--output "C:/CELFiles" --expname "CancerCompendium" --runGOElite yes
--returnPathways all
Analyzing RNA-Seq (RNASeq) data - BED files using default options
python AltAnalyze.py --species Mm --platform RNASeq --bedDir "C:/BEDFiles"
--groupdir "C:/BEDFiles/groups.CancerCompendium.txt"
--compdir "C:/BEDFiles/comps.CancerCompendium.txt" --output "C:/BEDFiles"
--expname "CancerCompendium"
Analyzing CEL files - Exon 1.0 array using default options
python AltAnalyze.py --species Mm --arraytype exon --celdir "C:/CELFiles"
--groupdir "C:/CELFiles/groups.CancerCompendium.txt"
--compdir "C:/CELFiles/comps.CancerCompendium.txt" --output "C:/CELFiles"
--expname "CancerCompendium"
Analyzing Filtered Expression file - RNA-Seq using custom options
python AltAnalyze.py --species Mm --platform RNASeq --filterdir "C:/BEDFiles"
--altpermutep 1 --altp 1 --altpermute yes --additionalAlgorithm none
--altmethod linearregres --altscore 2 --removeIntronOnlyJunctions yes
Analyzing CEL files - Exon 1.0 array using custom options
python AltAnalyze.py --species Hs --arraytype exon --celdir "C:/CELFiles"
--output "C:/CELFiles" --expname "CancerCompendium" --runGOElite no --dabgp 0.01
--rawexp 100 --avgallss yes --noxhyb yes --analyzeAllGroups "all groups"
--GEcutoff 4 --probetype core --altp 0.001 --altmethod FIRMA --altscore 8
--exportnormexp yes --runMiDAS no --ASfilter yes --mirmethod "two or more"
--calcNIp yes
Analyzing CEL files - HJAY array using custom options
python AltAnalyze.py --species Hs --arraytype junction --celdir "C:/CELFiles"
--output "C:/CELFiles" --expname "CancerCompendium" --runGOElite no --dabgp 0.01
--rawexp 100 --avgallss yes --noxhyb yes --analyzeAllGroups "all groups"
--GEcutoff 4 --probetype core --altp 0.001 --altmethod "linearregres" --altscore 8
--exportnormexp yes --runMiDAS no --ASfilter yes --mirmethod "two or more"
--calcNIp yes --additionalAlgorithm FIRMA --additionalScore 8
Analyzing Expression file - Gene 1.0 array using default options, without GO-Elite
python AltAnalyze.py --species Mm --arraytype gene
--expdir "C:/CELFiles/ExpressionInput/exp.CancerCompendium.txt"
--groupdir "C:/CELFiles/groups.CancerCompendium.txt"
--compdir "C:/CELFiles/comps.CancerCompendium.txt"
--statdir "C:/CELFiles/ExpressionInput/stats.CancerCompendium.txt"
--output "C:/CELFiles"
Analyzing Filtered Expression file - Exon 1.0 array using default options
python AltAnalyze.py --species Hs --arraytype exon
--filterdir "C:/CELFiles/Filtered/Hs_Exon_prostate_vs_lung.p5_average.txt"
--output "C:/CELFiles"
Annotate External Probe set results - Exon 1.0 array using default options
python AltAnalyze.py --species Rn --arraytype exon
--annotatedir "C:/JETTA_Results/Hs_tumor_progression.txt"
--output "C:/JETTA_Results" --runGOElite yes
Filter AltAnalyze results with predefined IDs using default options
python AltAnalyze.py --species Mm --arraytype gene --celdir "C:/CELFiles"
--output "C:/CELFiles" --expname "CancerCompendium" --returnAll yes
Run Lineage Profiler ONLY
python AltAnalyze.py --input "/Users/rma/tumors.txt" --runLineageProfiler yes
--vendor Affymetrix --platform "3'array" --species Mm
Run Hierarchical Clustering ONLY
python AltAnalyze.py --input "/Users/filtered/pluripotency.txt" --image hierarchical
--row_method average --column_method single --row_metric cosine
--column_metric euclidean --color_gradient red_white_blue --transpose False
Run Principal Component Analysis ONLY
python AltAnalyze.py --input "/Users/rma/tumors.txt" --image PCA
Return colored WikiPathways ONLY
python AltAnalyze.py --input /Users/test/input/differentiation.txt
--image WikiPathways --mod Ensembl --species Hs --wpid WP536
Run GO-Elite ONLY
python AltAnalyze.py --input "/Mm_sample/input_list_small" --runGOElite yes
--denom "/Mm_sample/denominator" --output "/Mm_sample" --mod Ensembl --species Mm
--returnPathways all
Operating System Example Folder Locations
-
PC:
"C:/CELFiles" -
Mac OSX:
"/root/user/admin/CELFiles" -
Linux:
"/hd3/home/admin/CELFiles"
Primary Analysis Variables
No default value for these variables is given and must be supplied by the user if running an analysis. For example, if analyzing CEL files directly in AltAnalyze, you must include the flags --species, --arraytype, --celdir, --expname, and --output, with corresponding values. Likewise, when analyzing an existing expression file you must include the flags --species, --arraytype, --expdir, and --output. Most of the variable values are file or folder locations. These variable values will differ based on the directory path of your files and operating system (e.g, linux has a distinct path structure than windows - see above examples). The variable name used in the AltAnalyze source code for each flag is indicated below.
Universally Required Variables
--arraytype: (aka --platform) long variable name “array_type”. No default value for this variable. Options are RNASeq, exon, gene, junction, AltMouse and “3’array”. This variable indicates the general array type correspond to the input CEL files or expression file. An example exon array is the Mouse Affymetrix Exon ST 1.0 array, an example gene array is the Mouse Affymetrix Gene ST 1.0 array and example 3’array is the Affymetrix Mouse 430 version 2.0 array. See Affymetrix website for array classifications.
--species: long variable name “cel_file_dir”. No default value for this variable. Species codes are provided for this variable (e.g., Hs, Mm, Rn). Additional species can be added through the graphic user interface.
--output: long variable name “output_dir”. No default value for this variable. Required for all analyses. This designates the directory which results will be saved to.
Analysis Specific Required Variables
--expname: long variable name “exp_name”. No default value for this variable. Required when analyzing CEL files. This provides a name for your dataset. This name must match any existing groups and comps files that already exist. The groups and comps file indicate which arrays correspond to which biological groups and which to compare. These files must exist in the designated output directory in the folder “ExpressionInput” with the names “groups.expname.txt” and “comps.expname.txt” where expname is the variable defined in this flag. Alternatively, the user can name their CEL files such that AltAnalyze can directly determine which group they are (e.g., wildtype-1.CEL, cancer-1.CEL, cancer-2.CEL). See Creating Groups and Comps Outside AltAnalyze for more information
--celdir: (aka --bedDir) long variable name “cel_file_dir”. No default value for this variable. Required when analyzing CEL files. This provides the path of the CEL files to analyze. These must all be in a single folder.
--expdir: long variable name “input_exp_file”. No default value for this variable. Required when analyzing a processed expression file. This provides the path of the expression file to analyze.
--statdir: long variable name “input_stats_file”. No default value for this variable. Optional when analyzing a processed expression file. This provides the path of the DABG p-value file for the designated expression file to analyze (see -expdir).
--filterdir: long variable name “input_filtered_dir”. No default value for this variable. Required when analyzing an AltAnalyze filtered expression file. This provides the path of the AltAnalyze filtered expression file to analyze.
--cdfdir: long variable name “input_cdf_file”. No default value for this variable. Required when directly processing some CEL file types. This variable corresponds to the location of the Affymetrix CDF or PGF annotation file for the analyzed array. If you are analyzing an exon, gene, junction, AltMouse or 3’arrays, AltAnalyze has default internet locations for which to download these files automatically, otherwise, you must download the compressed CDF file from the Affymetrix website (support), decompress it (e.g., WinZip) and reference it’s location on your hard-drive using this flag. If you are unsure whether AltAnalyze can automatically download this file, you can try to exclude this variable and see if annotations are included in your gene expression results file.
--csvdir: long variable name “input_annotation_file”. No default value for this variable. Required when analyzing some expression files or CEL file types. This variable corresponds to the location of the Affymetrix CSV annotation file for the analyzed array. If you are analyzing an exon, gene, junction, AltMouse or 3’arrays, AltAnalyze has default internet locations for which to download these files automatically, otherwise, you must download the compressed CSV file from the Affymetrix website (support), decompress it (e.g., WinZip) and reference it’s location on your hard-drive using this flag. If you are unsure whether AltAnalyze can automatically download this file, you can try to exclude this variable and see if annotations are included in your gene expression results file.
--annotatedir: long variable name “external_annotation_dir”. No default value for this variable. Required when annotating a list regulated probe sets produced outside of AltAnalyze. This variable corresponds to the location of the directory containing one or more probe set files. These files can be in the standard JETTA export format, or otherwise need to have probe set IDs in the first column. Optionally, these files can have an associated fold change and p-value (second and third columns), which will be reported in the results file.
--groupdir: long variable name “groups_file”. No default value for this variable. Location of an existing group file to be copied to the directory in which the expression file is located or will be saved to.
--compdir: long variable name “comps_file”. No default value for this variable. Location of an existing comps file to be copied to the directory in which the expression file is located or will be saved to.
Optional Analysis Variables
These variables are set as to default values when not selected. The default values are provided in the configurations text file in the Config directory of AltAnalyze (default-***.txt) and can be changed by editing in a spreadsheet program.
GO-Elite Analysis Variables
AltAnalyze can optionally subject differentially or alternatively expressed genes (AltAnalyze and user determined) to an over-representation analysis (ORA) along Gene Ontology (GO) and pathways (WikiPathways) using the program GO-Elite. GO-Elite is seamlessly integrated with AltAnalyze and thus can be run using default parameters either the graphic user interface or command line. To run GO-Elite using default parameters in command line mode, include the first flag below with the option yes.
--runGOElite: long variable name “run_GOElite”, default value for this variable: no. Used to indicate whether to run GO-Elite analysis following AltAnalyze. Indicating yes would prompt GO-Elite to run.
--mod: long variable name “mod”, default value for this variable: Ensembl. Primary gene system for Gene Ontology (GO) and Pathway analysis to link Affymetrix probe sets and other output IDs to. Alternative values: EntrezGene.
--elitepermut: long variable name “goelite_permutations”, default value for this variable: 2000. Number of permutation used by GO-Elite to calculate an over-representation p-value.
--method: long variable name “filter_method”, default value for this variable: z-score. Sorting method used by GO-Elite to compare and select the top score of related GO terms. Alternative values: “gene number”, combined.
--zscore: long variable name “z_threshold”, default value for this variable: 1.96. Z-score threshold used following over-representation analysis (ORA) for reported top scoring GO terms and pathways.
--elitepval: long variable name “p_val_threshold”, default value for this variable: 0.05. Permutation p-value threshold used ORA analysis for reported top scoring GO terms and pathways.
--dataToAnalyze: long variable name “resources_to_analyze”, default value for this variable: both. Indicates whether to perform ORA analysis on pathways, Gene Ontology terms or both. Alternative values: Pathways or Gene Ontology.
--num: long variable name “change_threshold”, default value for this variable: 3. The minimum number of genes regulated in the input gene list for a GO term or pathway after ORA, required for GO-Elite reporting.
--GEelitepval: long variable name “ge_pvalue_cutoffs”, default value for this variable: 0.05. The minimum t-test p-value threshold for differentially expressed genes required for analysis by GO-Elite.
--GEeliteptype: default is rawp. Indicates whether to run rawp or adjp (Benjamini-Hochberg) p-value for filtering.
--GEelitefold: long variable name “ge_fold_cutoffs”, default value for this variable: 2. The minimum fold change threshold for differentially expressed genes required for analysis by GO-Elite. Applied to any group comparisons designated by the user.
--ORAstat: long variable name “ORA_algorithm”, default for this variable: Fisher Exact Test. When the value is set to “Permute p-value”, a permutation p-value will be calculated using the default of provided number of permutations (--elitepermut). The adjusted p-value will be calculated from the selected type of ORAstat.
--additional: long variable name “additional_resources”, default for this variable: None. When the value is set to one of a valid resource or “all”, GO-Elite will download and incorporate that resource along with the default downloaded (WikiPathways and Gene Ontology). Additional resources currently include the options: “miRNA Targets”, ”GOSlim”, ”Disease Ontology”, ”Phenotype Ontology”, ”KEGG”, “Latest WikiPathways”, ”PathwayCommons”, ”Transcription Factor Targets”, ”Domains” and ”BioMarkers” (include quotes).
--denom: long variable name “denom_file_dir”, default for this variable: None. This is the folder location containing denominator IDs for corresponding input ID list(s). This variable is only supplied to AltAnalyze when independently using the GO-Elite function to analyze a directory of input IDs (--input) and a corresponding denominator ID list.
--returnPathways: long variable name “returnPathways”, default for this variable: None. When set equal to “yes” or “all”, will return all WikiPathways as colored PNG or PDF files (by default both) based on the input ID file data and over-representation results. Default value is “None”. When equal to “top5”, GO-Elite will only produce the top 5 (or other user entered number - e.g., "top10") ranking WikiPathways.
AltAnalyze Expression Filtering and Summarization
These variables are used to determine the format of the expression data being read into AltAnalyze, the output formats for the resulting gene expression data and filtering thresholds for expression values prior to alternative exon analysis. Since AltAnalyze can process both convention (3’array) as well as RNA-Seq data and splicing arrays (exon, gene, junction or AltMouse), different options are available based on the specific platform.
Universal Array Analysis Variables
--logexp: long variable name “expression_data_format”, default value for this variable: log for arrays and non-log for RNA-Seq. This is the format of the input expression data. If analyzing CEL files in AltAnalyze or in running RMA or GCRMA from another application, the output format of the expression data is log2 intensity values. If analyzing MAS5 expression data, this is non-log.
--inclraw: long variable name “include_raw_data”, default value for this variable: yes. When the value of this variable is no, all columns that contain the expression intensities for individual arrays are excluded from the results file. The remaining columns are calculated statistics (groups and comparison) and annotations.
--vendor: default is "Affymetrix". This variable can be set to "Other ID" when analyzing data from proteomics, metabolomics or other data not explicitly listed in the vendor/data-type menu. When entering "Other ID", also include the --platform as the specific ID system (e.g., PubChem).
RNASeq, Exon, Gene, Junction or AltMouse Platform Specific Variables
--dabgp: long variable name “dabg_p”, default value for this variable: 0.05. This p-value corresponds to the detection above background (DABG) value reported in the “stats.” file from AltAnalyze, generated along with RMA expression values. A mean p-value for each probe set for each of the compared biological groups with a value less than this threshold will be excluded, both biological groups don’t meet this threshold for a non-constitutive probe set or if one biological group does not meet this threshold for constitutive probe sets.
--rawexp: long variable name “expression_threshold”, default value for this variable: 70 for microarrays and 2 for RNASeq reads. For Affymetrix arrays, this value is the non-log RMA average intensity threshold for a biological group required for inclusion of a probe set. The same rules as the --dabgp apply to this threshold accept that values below this threshold are excluded when the above rules are not met.
--avgallss: long variable name “avg_all_for_ss”, default value for this variable: no. For RNA-Seq analyses, default is yes. Indicating yes, will force AltAnalyze to use all exon aligning probe sets or RNA-Seq reads rather than only features that align to predicted constitutive exons for gene expression determination. This option applies to both the gene expression export file and to the alternative exon analyses.
--runalt: long variable name “perform_alt_analysis”, default value for this variable: yes. Designating no for this variable will instruct AltAnalyze to only run the gene expression analysis portion of the program, but not the alternative exon analysis portion.
--groupStat: default is "moderated t-test". To designate an alternate statistic, this variable can be set to one of the following options: "paired t-test", "Kolmogorov Smirnov", "Mann Whitney U", "Rank Sums".
AltAnalyze Alternative Exon Statistics, Filtering and Summarization
Universal Array Analysis Variables
--altmethod: long variable name “analysis_method”, default value for this variable: splicing-index (exon and gene) and ASPIRE (RNASeq, junction and AltMouse). For exon, gene and junction arrays, the option FIRMA is also available and for RNASeq, junction and AltMouse platforms the option linearregres is available.
--altp: long variable name “p_threshold”, default value for this variable: 0.05. This variable is the p-value threshold for reporting alternative exons. This variable applies to both the MiDAS and splicing-index p-values.
--probetype: long variable name “filter_probe set_types”, default value for this variable: core (exon and gene) and all (junction and AltMouse). This is the class of probe sets to be examined by the alternative exon analysis. Other options include, extended and full (exon and gene) and “exons-only”, “junctions-only”, “combined-junctions” (RNASeq, junction and AltMouse).
--altscore: long variable name “alt_exon_fold_variable”, default value for this variable: 2 (splicing-index) and 0.2 (ASPIRE).This is the corresponding threshold for the default algorithms listed under --altmethod.
--GEcutoff: long variable name “gene_expression_cutoff”, default value for this variable: 3. This value is the non-log gene expression threshold applied to the change in gene expression (fold) between the two compared biological groups. If a fold change for a gene is greater than this threshold it is not reported among the results, since gene expression regulation may interfere with detection of alternative splicing.
--analyzeAllGroups: long variable name “analyze_all_conditions”, default value for this variable: pairwise. This variable indicates whether to only perform psiteifalternative exon analyses (between two groups) or to analyze all groups, without specifying specific comparisons. Other options are “all groups” and both.
--altpermutep: long variable name “permute_p_threshold”, default value for this variable: 0.05. This is the permutation p-value threshold applied to AltMouse array analyses when generating permutation based alternative exon p-values. Alternative exon p-values can be applied to either ASPIRE or linregress analyses.
--altpermute: long variable name “perform_permutation_analysis”, default value for this variable: yes. This option directs AltAnalyze to perform the alternative exon p-value analysis for the AltMouse array (see --altpermutep).
--exportnormexp: long variable name “export_splice_index_values”, default value for this variable: no. This option directs AltAnalyze to export the normalized intensity expression values (feature expression/constitutive expression) for all analyzed features (probe sets or RNA-Seq reads) rather than perform the typical AltAnalyze analysis when its value is yes. For junction-sensitive platforms, rather than exporting the normalized intensities, the ratio of normalized intensities for the two reciprocal-junctions are exported (pNI1/pNI2). This step can be useful for analysis of exon array data outside of AltAnalyze and comparison of alternative exon profiles for many biological groups (e.g., expression clustering).
--runMiDAS: long variable name “run_MiDAS”, default value for this variable: yes. This option directs AltAnalyze to calculate and filter alternative exon results based on the MiDAS p-value calculated using the program Affymetrix Power Tools.
--calcNIp: long variable name “calculate_normIntensity_p”, default value for this variable: yes. This option directs AltAnalyze to filter alternative exon results based on the t-test p-value obtained by comparing either the normalized intensities for the array groups examined (e.g., control and experimental) (splicing-index) or a t-test p-value obtained by comparing the FIRMA scores for the arrays in the two compared groups.
--mirmethod: long variable name “microRNA_prediction_method”, default value for this variable: one. This option directs AltAnalyze to return any microRNA binding site predictions (default) or those that are substantiated by multiple databases (two or more).
--ASfilter: long variable name “filter_for_AS”, default value for this variable: no. This option directs AltAnalyze to only analyze probe sets or RNA-Seq reads for alternative expression that have an alternative-splicing annotation (e.g., mutually-exclusive, trans-splicing, cassette-exon, alt-5’, alt-3’, intron-retention), when set equal to yes.
--returnAll: long variable name “return_all”, default value for this variable is no. When set to yes, returns all un-filtered alternative exon results by setting all associated filtering parameters to the lowest stringency values. This is equivalent to providing the following flags: --dabgp 1 --rawexp 1 --altp 1 --probetype full --altscore 1 --GEcutoff 10000. Since this option will output all alternative exon scores for all Ensembl annotated junctions or probe sets, the results file will be exceptionally large (>500,000 lines), unless the user has saved previously run alternative exon results (e.g., MADS) to the directory “AltDatabase/filtering” in the AltAnalyze program directory, with a name that matches the analyzed comparison. For example, if the user has a list of 2,000 MADS regulated probe sets for cortex versus cerebellum, then the MADS results should be saved to “AltDatabase/filtering” with the name “Cortex_vs_Cerebellum.txt” and in AltAnalyze the CEL file groups should be named Cortex and Cerebellum and the comparison should be Cortex versus Cerebellum. When the filename for a file in the “filtering” directory is contained within the comparison filename (ignoring “.txt”), only these AltAnalyze IDs or probe sets will be selected when exporting the results. This analysis will produce a results file with all AltAnalyze statistics (default or custom) for just the selected features, independent of the value of each statistic.
--additionalAlgorithm: long variable name “additional_algorithms”, default value for this variable: "splicing-index". For Affymetrix arrays, setting this flag equal to FIRMA changes the individual probe set analysis algorithm from splicing-index to FIRMA for junction arrays. This method is applied to RNA-Seq data and junction arrays following reciprocal-junction analysis (e.g., ASPIRE) in a second run. To exclude this feature, set variable equal to none.
--additionalScore: long variable name “additional_score”, default value for this variable: 2. Setting this flag equal to another numeric value (range 1 to infinity) changes the non-log fold change for the additional_algorithms.
--removeIntronOnlyJunctions: long variable name “remove_intronic_junctions”, default value for this variable: no. Indicates whether to remove junctions where both splice sites align to outside of annotated exons. Setting this value to yes will remove these putative junctions prior to analysis.
--normCounts: long variable name “normalize_feature_expression”, default value for this variable: none. Indicates whether to normalize exon and/or junctions counts using the methods RPKM or quantile normalization. Setting this value to none will use the original counts for gene expression and alternative exon analyses.
--buildExonExportFile: default is no. Indicates whether to halt an RNA-Seq analysis after reading in the junction.bed files and export a new file with the suffix exon.bed in the folder BAMtoBED. This file is used as described here to obtain exon.bed files for all experimental BAM files. Setting this value to yes will use the original counts for gene expression and alternative exon analyses.
--groupStat: default is “moderated t-test”. Indicates the algorithm to employ for all pairwise group comparisons in AltAnalyze. Other options include: “paired t-test”, “Kolmogorov Smirnov”, “Mann Whitney U” and “Rank Sums”.
--exonRPKM: default is 0.5. Numerical filter used to threshold exons as expressed when using RPKM normalization for mean RPKM values for each biological group. At least one biological group for a pairwise comparison must meet this threshold to be included in further analyses. Increasing values of this filter will increase the stringency of exon/gene expression.
--geneRPKM: default is 1. Numerical filter used to threshold exons as expressed when using RPKM normalization for mean RPKM values for each biological group. At least one biological group for a pairwise comparison must meet this threshold to be included in further analyses. Increasing values of this filter will increase the stringency of exon/gene expression.
--exonExp: default is 10. Numerical filter used to threshold genes or exons as expressed based on the mean read counts values for each biological group. At least one biological group for a pairwise comparison must meet this threshold to be included in further analyses. Increasing values of this filter will increase the stringency of exon/gene expression.
AltAnalyze Database Updates
Universal Array Analysis Variables
--update: long variable name “update_method”, default value for this variable: empty. Setting this flag equal to Official, without specifying a version, will download the most up-to-date database for that species. Other options here are used internally by AltAnalyze.org for building each new database. See the method “commandLineRun” in AltAnalyze.py for more details.
--version: long variable name “ensembl_version”, default value for this variable: current. Setting this flag equal to a specific Ensembl version name (e.g. EnsMart49) supported by AltAnalyze will download that specific version for the selected species, while setting this to current will download the current version.
--specificArray: default is none. Indicates a sub-type of a particular array platform (e.g., junction - HJAY or hGlue) when building the database. This variable only needs to be set when currently building the hGlue junction database (see here).
--ignoreBuiltSpecies: default is no. Indicating yes will only build species databases for species without already built species directories. This is used during internal database release building to simultaneously build multiple species databases.
Additional Analysis, Quality Control and Visualization Options
In addition to the core AltAnalyze workflows (e.g., normalization, gene expression summarization, evaluation of alternative splicing), additional options are available to evaluate the quality of the input data (quality control or QC), evaluate associated cell types and tissues present in each biological sample (Lineage Profiler), cluster samples or genes based on overall similarity (expression clustering) and view regulation data on biological pathways (WikiPathways). These options can be run as apart of the above workflows or often independently using existing AltAnalyze results or input from other programs.
--outputQCPlots: long variable name “visualize_qc_results”, default value for this variable: yes. Instructs AltAnalyze to calculate various QC measures specific to the data type analyzed (e.g., exon array, RNASeq) when a core workflow is run. This will include hierarchical clustering and PCA plots for genes considered to be differentially expressed (see --GEelitefold and --GEelitepval). Outputs various QC output plots (PNG and PDF) to the folder “DataPlots” in the user defined output directory. If run from python source code, requires installation of Scipy, Numpy and Matplotlib.
--runLineageProfiler: long variable name “run_lineage_profiler”, default value for this variable: yes. Instructs AltAnalyze to calculate Pearson correlation coefficients for each analyzed user sample relative to all cell types and tissues in the BioMarker database. Resulting z-scores for calculated from the coefficients are automatically visualized on a WikiPathways Lineage network and are hierarchically clustered. Outputs various tables to the folder ExpressionOutput and plots (PNG and PDF) to the folder “DataPlots” in the user defined output directory. Can be run as apart of an existing workflow or independently with the option --input. If run from python source code, requires installation of suds, Scipy, Numpy and Matplotlib.
--input: long variable name “input_file_dir”, default value for this variable: None. Including this option indicates that the user is referencing an expression file location that is supplied outside of a normal AltAnalyze workflow. Example analyses include only performing hierarchical clustering, PCA or GO-Elite.
--image: long variable name “image_export”, default value for this variable: None. Including this option with any of the variables: WikiPathways, hierarchical, PCA, along with an input expression file location (--input), will prompt creation and export of the associated visualization files. These analyses are outside of the typical AltAnalyze workflows, only requiring the designated input file. If run from python source code, requires installation of suds, Scipy, Numpy and Matplotlib.
Hierarchical Clustering Variables
--row_method: long variable name “row_method”, default value for this variable: average. Indicates the cluster metric to be applied to rows. Other options include: average, single, complete, weighted and None. None will result in no row clustering.
--column_method: long variable name “column_method”, default value for this variable: single. Indicates the cluster metric to be applied to columns. These options are the same as --row_method.
--row_metric: long variable name “row_metric”, default value for this variable: cosine. Indicates the cluster distance metric to be applied to rows. Other options include: braycurtis, canberra, chebyshev, cityblock, correlation, cosine, dice, euclidean, hamming, jaccard, kulsinski, mahalanobis, matching, minkowski, rogerstanimoto, russellrao, seuclidean, sokalmichener, sokalsneath, sqeuclidean and yule (not all may work). If the input metric fails during the analysis (unknown issue with Numpy), euclidean will be used instead.
--column_metric: long variable name “column_metric”, default value for this variable: euclidean. Indicates the cluster distance metric to be applied to rows. These options are the same as --row_metric.
--color_gradient: long variable name “image_export”, default value for this variable: red_white_blue. Indicates the color gradient to be used for visualization as up-null-down. Other options include red_black_sky, red_black_blue, red_black_green, yellow_black_blue, green_white_purple, coolwarm and seismic.
--transpose: long variable name “transpose”, default value for this variable: False. Will transpose the matrix of columns and rows prior to clustering, when set to True.
AltAnalyze Analysis Options
There are a number of analysis options provided through the AltAnalyze interface. This section provides an overview of these options for the different compatible analyses (gene expression arrays, exon arrays, junction arrays and RNA-Seq data). For new users, we recommend first running the program with the pre-set defaults and then modifying the options as necessary.
Selecting the Platform and Species
When beginning AltAnalyze, the user can select from a variety of species and platform types. Only array manufacturers and array types supported for each downloaded species will be displayed along with support for RNA-Seq analysis. When multiple gene database versions are installed, a drop-down box at the top of this screen will appear that allows the user to select different gene database versions. These gene databases include all resources necessary for gene annotation, alternative exon analysis (where applicable) and Gene Ontology and pathway analysis. Expression normalization, summarization, annotation and statistical analysis options are available for all input data types (e.g., microarray, RNASeq, proteomics, metabolomics data). At the bottom of this interface is a check-box that the user can select to download updated species gene databases, which will bring-up the database downloader window.
Selecting the RNA-Seq Analysis Method
Similar to microarray analysis options (see below), users can choose to analyze; 1) FASTQ files (gene expression only), 2) BAM/BED/TAB/TCGA junction files, 3) an already built RNA-Seq expression file or 4) an AltAnalyze filtered RNA-Seq expression file for RNA-Seq data.
Option 1 uses the embedded software Kallisto, which is automatically called to produce pseudoaligned and quantified transcript expression values from the user supplied single-end or paired-end FASTQ files. The Kallisto k-mer transcript database is built using annotations from Ensembl which AltAnalyze downloads automatically, corresponding to the correct version of the Ensembl (e.g., Ensembl 72). Transcript-level expression values (TPMs) for mRNAs with experimental evidence are summed at the gene-level and saved with the prefix exp. to the folder ExpressionInput. A summary file indicates the estimated read depth and percentage alignment of each FASTQ pair. Results from this analysis are immediately processed using the user-defined AltAnalyze workflow options. More information on this workflow can be found here.
AltAnalyze Since options 3 and 4 produce files from option 2, users will want to begin by loading a directory of RNA-Seq counts (junction and/or exon) from their alignment analysis results (BAM files or tabular coordinate result files). Various programs can be used to produce the BAM or junction .bed format files that are used as AltAnalyze input. These include HMMSplicer and TopHat. Exon .bed files are produced using BAM files and an AltAnalyze-produced input exon coordinate BED file with BEDTools (see Building a Dataset Exon Database for BEDTools). Exon .bed and even junction .bed files are now automatically generated from supplied BAM files through AltAnalyze rather than deriving these through BEDTools, although users can still derive these via BEDTools if they prefer. The junction and exon BED files consist of junction splice-site coordinates along with the number of reads from a sequencing run that correspond to that junction.
BED and TAB File Summarization
After loading junction BED files, AltAnalyze will like link each junction to an Ensembl gene and known splice-sites based on the provided genomic coordinates. When novel splice-sites are encountered, AltAnalyze will create a novel junction annotation for the splice-site (5’ or 3’). In some cases, the splice-sites may be present in two different genes, indicating trans-splicing. The number of known-splice sites, novel splice sites and trans-splicing junctions will be reported upon import and in the log file. The resulting file, saved to the folder ExpressionInput/exp.NameYouEnter.txt, will contain unique identifiers indicating the Ensembl gene and associated exon-junction (e.g., E13.1-E14.1), indicating the exon block (e.g., “E13”) and exon region (e.g., “.1”) that the splice site positions aligns to. When the splice-site is novel it will be annotated as aligning to the corresponding exon, intron or UTR region with the additional notation “_position”, where position is the genomic splice-site coordinate, following the exon region (e.g., E13.1_1000347, I13.1_1000532, U15.1_1001023). If exon .bed files are present in this same directory have the same prefix name (e.g., sample1__exon.bed and sample1__junction.bed), the exon coordinates will be matched to AltAnalyze annotated exon and intron regions (Ensembl/UCSC) and novel exon regions inferred from the junction BED locations. Both junction expression and exon expression will be written to the same file with the prefix “exp.”. This file can subsequently be loaded as option 2 (“Process Expression File”). The same process is applied to .tab files from BioScope, accept that exon and junction count files are produced simultaneously and output to different format files.
Selecting the Microarray Analysis Method
After the user has selected the species of interest, they must choose what type of data they will next be analyzing. Data can consist of; 1) Affymetrix CEL files, 2) an already processed expression text files, 3) properly formatted and filtered AltAnalyze expression input text file or 4) restricted list of probe sets to be directly analyzed. If beginning with Affymetrix CEL files all three of these file types are produced in series (see following section) and automatically processed without any user intervention. If all CEL files from your study already been previously in AltAnalyze or in another program, the user can load this file be selecting the option “expression file” and choosing this text file from your computer. This file needs to contain data from arrays corresponding to at least two biological groups. Users may wish to re-analyze these files to change their expression filtering parameters to be more or less stringent. For the two or more biological groups (see how to define in Figure 2.8), AltAnalyze will segregate the raw data based on the user-defined pairwise group comparisons and filter the containing probe sets based on whether they match the user-defined thresholds for inclusion and are associated with Ensembl genes (see Expression Analysis Parameters). These files will be saved to the folder “AltExpression” in the user-defined output directory. These files can be later selected by choosing the option “AltAnalyze filtered”, if the user wishes to re-run or use different AltAnalyze alternative exon analysis options (see below section: Alternative Exon Analysis Parameters).
CEL File Summarization
CEL files are one of the file types produced after scanning an Affymetrix microarray. The CEL file is produced automatically from the DAT file (an image file, similar to a JPEG), by the Affymetrix software by overlaying a grid over the microarray florescent image and assigning a numeric value to each cell or probe. From this file, expression values for each probe set can be calculated and normalized for all arrays in the study using various algorithms.
When choosing to analyze CEL files in AltAnalyze, the user will be prompted to identify the folder containing the CEL files and the folder in which to save these other results to. The user will also need to assign a name to the dataset. These CEL files will be summarized using the RMA algorithm using the program Affymetrix Power Tools (APT). The APT C++ module “apt-probeset-summarize” is directly called by AltAnalyze when running AltAnalyze on a Mac, PC or Linux operating system. Unlike some other applications, APT is packaged with AltAnalyze and thus does not require separate installation. However, because it is a separate application there may be unknown compatibility issues that exist, depending on your specific system configuration and account privileges. For human and mouse exon arrays, AltAnalyze also allows for the masking of probes with cross-hybridization potential, prior to running RMA. This is performed through an experimental APT function (--kill-list), masking probes that are indicated in files produced for the MADS application[Xing2008] that cross-hybridize to an off-target transcript within 3bp mismatches and a person correlation coefficient > 0.55, as per the MADS recommendations. The probes with cross-hybridiation potential are indicated in the AltAnalyze directory “AltDatabase/Hs/exon/Hs_probes_to_remove.txt”.
APT requires the presence of a library file(s) specific for that array. AltAnalyze will automatically determine the array type and can install these files if the user wishes (currently most human, rat and mouse arrays supported). If AltAnalyze does not recognize the specific array type or the user chooses to download these files themselves, they will need to select the appropriate files when prompted in AltAnalyze. For exon, gene and junction arrays, a PGF, CLF and antigenomic BGP file are required. These files will be automatically downloaded and installed if the user selects “Download” when prompted. For the AltMouse and 3’arrays, the appropriate CDF file will be downloaded. In addition to these library files, a NetAffx CSV annotation file will be downloaded that allows for addition of gene annotations (non-exon arrays) and Gene Ontology pathway annotations (all arrays). Once installed, AltAnalyze will recognize these files and automatically use them for all future analyses. Once the user selects the appropriate directories and files, the user will be prompted to select the remaining options in AltAnalyze, before APT is run. Once run, a tab-delimited text expression file will be produced for all probe sets on the array and a detection-above background (DABG) p-value file (not applicable to AltMouse or 3’arrays).
Loading a Processed Expression File
If performing an RNA-Seq analysis, this is the file produced immediately after loading and aligning the junctions to exons, introns and UTR regions. For Affymetrix arrays, if CEL files are processed outside of AltAnalyze, the user must save the resulting expression text file in tab-delimited format. It is all right if the first rows in the file have run information as long as they are preceded by a pound sign (#).
Expression Analysis Parameters
The options presented in this interface (Figure 2.6) allow the user to determine what fields are present in the gene expression output file, what scale the data is in (e.g. logarithmic), which RNA-Seq reads or probe sets (aka features) to use when calculating gene expression and how to filter features for subsequent analyses.
-
Perform an alternative exon analysis - Selecting the option “just expression” will halt the analysis after the gene expression result file has been written, such that no splicing analysis is performed. This option is only available for splicing-sensitive platforms.
-
Expression data format - Indicates the format in which the normalized expression values or counts have been written. When CEL files have been processed by AltAnalyze, ExpressionConsole, APT, RMAExpress or through R, the file format will be logarithmic base 2 (log). The default format of RNA-seq counts is non-log. If the user designates “non-log”, then expression values will be log base 2 (log2) transformed prior to analysis.
-
Determine gene expression levels using - For splicing RNA-Seq and sensitive microarrays, the user has the choice to alter the way in which gene expression values are calculated and how to filter their feature-level expression files prior to alternative exon analysis. When “core” features are selected for this option, all core features (Affymetrix core annotated and any exon aligning feature) linked to a unique gene will be used to calculate a measure of gene expression by taking the mean expression of all associated feature values. When the “constitutive” is selected, only those features that have been annotated as constitutive or common to the most isoforms will be used for gene expression calculation. In either case, only features with at least one array possessing a DABG p-value less than the user threshold (Affymetrix only) will be retained (if a DABG p-value file is present). In order to exclude this threshold, set the minimum DABG p-value equal to 1.
-
Include replicate experimental values in the export - Instructs AltAnalyze whether to include the expression values associated with each BED or CEL file in the output file. If not selected only the mean expression value of all BED or CEL files for each biological group will be written.
-
Remove probesets with a DABG p-value above - When a DABG file has been produced (default when summarizing CEL files with AltAnalyze for exon-arrays), this option is applied. The default DABG p-value cutoff is p<0.05. This will filter out any non-constitutive probe set that has a mean DABG p>0.05 for both compared biological groups. For probe sets used in determining gene expression levels, both biological groups must have a DABG p < user-value. In order to exclude this option, you can remove the DABG file (contains the prefix “stats.”), or set this value equal to 1.
-
Remove probesets/reads expressed below (non-log) - Filters can be applied to both Affymetrix array data and RNA-Seq datasets to exclude non-expressed probesets, genes, exons or junctions. In all cases, the expression of a feature is examined (non-log value) to see if that feature meets the indicated threshold. If not, it is excluded or considered non-expressed. This can be critical for comparisons where neither condition demonstrates gene or exon expression and hence can be considered an artifact. To exclude this option, set the default value to 1 for probeset or read count thresholds and 0 for RPKM filters.
-
Comparison group test statistic - Allows the user to calculate a p-value for gene expression and splicing analyses based on different tests (e.g., paired versus unpaired t-test, rank sum, Mann Whitney, Kolmogorov Smirnov). These test apply to two sample group comparisons, whereas any multi-group comparison analyses rely only on an f-test statistic. For unpaired t-test, the f-test statistic is also used. These tests are provided through a module of the open-source statistical package Salstat.
-
Perform expression clustering and visual QC - This option will automatically generate various quality control plots and hierarchical clustering heatmaps based on the user input dataset analyzed. Basic quality control metrics include the: 1) distribution of normalized log2 expression values, 2) raw signal intensities (Affymetrix - prior to APT), 3) deviation of residuals from the mean (Affymetrix - post RMA), 4) Feature-level (exon, junction, intron) expression box-plots (RNA-Seq) and 5) the total expression of each feature (RNA-Seq) for all analyzed samples. Principal component analysis and hierarchical clustering are also applied to all significantly regulated genes (default or user-defined criterion). Hierarchical clustering is also applied to genes with outlier expression values to identify poor-replicating samples. When this option is selected, the results will be available as PDF and PNG in the folder DataPlots and from the GUI once the analysis is finished (Figure 2.9 Top). When run from source-code, requires installation of MatPlotlib and Numpy.
-
Perform cell profiling with LineageProfiler - Lineage Profiler is a novel tool designed to analyze and visualize the cellular composition of supplied RNA profiles. Only RNA profiling data with gene expression values (e.g., Affymetrix and RNA-Seq), as opposed to folds only, are currently supported. Lineage Profiler produced pearson correlation coefficients and associated Z scores for each sample analyzed. These Z scores are visualized as a hierarchically clustered heatmap for all samples and as a comprehensive lineage network for the biological groups (Figure 2.17). When running from source-code this tool requires the python libraries lxml, MatPlotLib and Numpy for results visualization and is optimized to run with Scipy.
-
Perform ontologies and pathways with GO-Elite - Choosing “decide later” will allow the user to view the GO-Elite pathway and Gene Ontology over-representation analysis options after the main gene expression and/or alternative exon analysis is run. This will prompt a separate status window and results summary window displaying over-representation statistics for pathway analysis. If the option “run immediately” is selected, GO-Elite will run right away without a separate window. Please note, GO-Elite analysis can take up to an hour per criterion when using the default parameters, when analyzing all possible gene-sets, pathways and ontologies. For this reason, multithreading has been implemented in GO-Elite version 1.2.6 and greater. More details can be found here.
Alternative Exon Analysis Parameters
The options presented in this menu (Figure 2.7) instruct AltAnalyze what statistical methods to use when determining alternative exon expression, which features to select for analysis, what domain-level and miR-BS analyses to perform and what additional values to export for analyses in other tools. Details on each analysis algorithm are covered in detail in Algorithm Descriptions.
-
Select the alternative exon algorithm - For exon and gene arrays, the splicing index and FIRMA methods are available and for RNA-Seq and junction arrays the ASPIRE and linear regression methods are available. RNA-Seq and junction analyses can also include single junction analyses (e.g., Splicing-index, MiDAS), following the reciprocal junction analysis. These methods are used to calculate an alternative exon score, relative to gene expression levels. The default value for splicing-index and FIRMA analyses is 2, indicating that an adjusted expression difference greater than two fold (up- or down-regulated) is required for the probe set to be reported. Based on the algorithm, different values and scales will apply. For junction analyses, the ASPIRE algorithm default cutoff is 0.2, whereas the linear-regression algorithm is 2. For linear-regression (linearregres), a minimum value of 2 will select any linear-regression fold greater than 2 (result folds are reported in log 2 scale, however), up- or down-regulated, whereas ASPIRE’s scores ranging from -1 to 1. See Algorithm Descriptions for more details.
-
Minimum alternative exon score - This value will vary based on the alternative exon analysis method chosen (see above options).
-
Max MiDAS/normalized intensity p-value - This is the p-value cutoff applied to MiDAS and splicing-index or FIRMA ttest p-values for single exon/junction analyses. Currently, the user cannot set different p-value thresholds for these two statistics. More on MiDAS can be found below and in Algorithm Descriptions.
-
Select probesets to include - This option is used to increase or decrease the stringency of the analysis. In particular, this option allows the user to restrict what type of features are to be used to calculate an alternative exon score. In the case of junction analyses, this option includes the ability to merge the expression values of junctions/exons that measure the same differential inclusion of an exon (combined-junctions). For exon and gene arrays, there are three options, “core”, “extended” and “full”. Although these are the same probe set class names used by Affymetrix to group probe sets, AltAnalyze uses a modification of these annotations. Specifically, probe sets with the core annotation include all Affymetrix core probe sets that specifically overlap with a single Ensembl gene [Hubbard2007] (based on genomic position) along with any probe set that overlaps with an Ensembl or UCSC exon [Karolchik2008]. Likewise, extended and full probe sets are those remaining probe sets that also align to a single Ensembl gene, with the Affymetrix extended or full annotation. For RNA-Seq and junction arrays, options include “all”, “exons-only”, “junctions-only”, “combined-junctions”.
-
Maximum absolute gene-expression change - This value indicates maximum gene expression fold change (non-log, up- or down-regulated) that is allowed for a gene to be reported as alternatively regulated. The default is 3-fold, up or down-regulated. This filter is used with assumption that alternative splicing is a less critical factor when a gene is highly differentially expressed.
-
Perform permutation analysis - (Junction Analyses Only) This analysis reports a p-value that represents the likelihood of the observed alternative exon score occurring by change, after randomizing the expression values of all samples.
-
Maximum reciprocal-junction permutatep - (Junction Analyses Only) This p-value cutoff applies to the permutation based alternative exon score p-values when performing ASPIRE or linearregres (see Algorithm Descriptions).
-
Export all normalized intensities - This option can be used to compare alternative exon scores prior to filtering for biological multiple comparisons, outside of AltAnalyze. For example, if comparing multiple tissues, the user may wish to export all normalized intensities (feature non-log expression divided by gene-expression) for all tissue comparisons. The results will be stored to the AltResults/RawSpliceData folder in the user-defined output-directory. For junction analyses, the ratio of the normalized intensities (NI) is reported for the two reciprocal-junctions (j1 and j2).
-
Calculate MiDAS p-values - This statistic is analogous to the ttest p-value calculated during the splicing-index analysis (see Algorithm Descriptions for more details). If not selected, then only the splicing-index or FIRMA (depending on the user selection) fold and p-value will be used to filter alternative exon results.
-
Calculate normalized intensity p-values - Indicates whether to calculate the splicing-index or FIRMA ttest p-values and filter using the above threshold.
-
Filter results for predicted AS - This option instructs AltAnalyze to only include regulated exons in the output that have been assigned a valid splicing annotation (e.g., alternative-cassette exon) provided by AltAnalyze. These annotations exclude exons with no annotations or those with only an alternative N-terminal exon or alternative promoter annotation.
-
Align feature to protein domains using - This option is used to restrict the annotation source for domain/motif over-representation analysis. If “direct-alignment” is chosen, only those features that overlap with the genomic coordinates of a protein domains/motif will be included in the over-representation analysis, otherwise, the inferred method is used (see Algorithm Descriptions for more details).
-
Number of algorithms required for miRNA binding site reporting - This option is used to filter out miR-BS predictions that only occur in one of the four miR-BS databases examined. For more miR database information, see Extracting microRNA Binding Annotations Overview.
-
Type of group comparison to perform - This option indicates whether to only perform pairwise alternative exon analyses (between two groups) or to analyze all groups, without specifying specific comparisons.
Overview of Analysis Results
AltAnalyze will produce three sets of results:
-
Gene expression (GE)
-
Alternative exon
-
Diagnostic and exploratory visualization
These files are all saved to the user-defined output directory and can be explored through the use of a spreadsheet data viewer, such as Microsoft Excel or OpenOffice, and a PDF viewer. Issues reading these spreadsheets may occur on non-US Windows configurations that (e.g., improper processing of numbers of with decimals). Additional information on the statistical methods used and source of annotations can be found in Algorithms.
Gene Expression Summary Data
There are five primary GE summary result files produced by AltAnalyze. These files contain raw data, summary statistics and/or comprehensive annotations:
-
DATASET file - Gene annotations, comparison and ANOVA statistics, raw expression values and counts (saved to ExpressionOutput).
-
GenMAPP file - Comparison statistics (saved to ExpressionOutput).
-
Summary statistics file - Overview statistics for protein and non-coding genes, up- and down-regulated counts and microRNA binding site statistics (saved to ExpressionOutput).
-
Clustering input file - Contains all differentially expressed genes based on user comparisons (saved to ExpressionOutput/Clustering).
-
GO-Elite input files - Lists of differentially expressed genes as input for pathway over-representation (saved to the folder GO-Elite).
The first file is a complete dataset summary file with the prefix “DATASET-” followed by the user-defined dataset name containing all array expression values (gene-level for RNA-Seq and tiling-arrays), calculated group statistics (mean expression, folds, raw and adjusted t-test and f-test p-values) and gene annotations (e.g., gene symbol, description, Gene Ontology, pathway and some custom groups, genomic location, protein coding potential, miRNA binding sites). For microRNA arrays, other annotations from the Affymetrix annotation CSV will replace these (user supplied). For RNA-Seq and tiling arrays, the gene expression values are derived from either RNA-Seq reads or probe sets most informative for transcription (known exons or constitutive) (see Expression Analysis Parameters). For RNA-Seq, if only junctions are present, then constitutive junctions or known junctions will be used, however, if exon reads are present, these will be used over junction reads. Constitutive features are determined by finding discrete exon regions that are common to the most mRNA transcripts (Ensembl and UCSC) for all transcripts used in the AltAnalyze database build (see Building AltAnalyze Annotation Files). For junction arrays (hGlue, HJAY, HTA2.0 and MJAY), both constitutive exon aligning probe sets and constitutive junction aligning junction probe sets (most common junctions in mRNAs) are used, whereas for RNA-Seq, constitutive exons are used. When one or more gene expression reporting probe sets have DABG p-values with at least one biological group with a mean value below the user defined threshold, these probe sets will be used to calculate gene expression, otherwise, all gene expression reporting probe sets will be used.
The second file, with the prefix “GenMAPP-”, contains a subset of columns from the dataset summary file for import into GenMAPP[Salomonis2007] or PathVisio[vanIersel2008]. This file has the prefix “GenMAPP” and excludes all gene annotations and individual sample expression values. This text file can be imported into these programs to create criterion and color the associated results on pathways (see Performing Pathway Analysis Downstream of AltAnalyze).
The third file, with the prefix “SUMMARY-”, contains overview statistics for that dataset. These results are divided in to Ensembl protein coding and non-coding genes. Counts for up- and down-regulated genes are provided separately. Genes called “expressed” are most relevant for RNA-Seq analyses, where raw counts incremented by 1 for fold calculation purposes (gene-level). A microRNA count is provided for a single miRNA (miR-1 by default). To change this, users must manually edit the file ExpressionBuilder.py (exportGeneRegulationSummary function).
The fourth file, with the prefix “SampleLogFolds-“, provides all log2 fold changes relative to the mean expression of all samples in the dataset for all “regulated” genes. Genes included in this file are those that have greater than 2 fold (up or down) change in gene expression and comparison statistic p < 0.05 for any user indicated comparison. To change these defaults, the user must currently select “run immediately” for the GO-Elite analysis option and change the associated defaults. This file will be used for (A) hierarchical clustering and (B) principal component analysis, when these options are selected.
The fifth file type, saved to the folder “GO-Elite/input” are differentially or alternative expressed genes and summary statistics for all comparisons. These files are primarily used as input for performing ontology, pathway or gene-set enrichment analysis using the GO-Elite option. This analysis can be run along with the default AltAnalyze workflows, immediately afterwards, or independently using the Additional Analyses menu (Pathway Enrichment option). These lists can also be used for Pathway Visualization also from the Additional Analyses menu. These files contain all differentially expressed genes (prefix GE.), alternative regulated (prefix AS.), as well as up and downregulated genes (suffix -upregulated or -downregulated). The IDs listed will be either the primary microarray identifier or an Ensembl gene ID, based on the platform analyzed. Also included are the log2-fold changes and p-values associated with the indicated comparisons. These files may also be of use in external analysis programs.
Alternative Exon Summary Data
These results are produced from all features (RNA-Seq reads or probe sets) that may suggest alternative splicing, alterative promoter regulation, or any other variation relative to the gene expression for that gene (derived from comparisons file). When the user chooses to either analyze all groups rather than just pairwise comparisons or both, the same output files will be produced but report MiDAS p-values comparing all conditions and the maximum possible splicing index fold between all conditions (see Algorithm Descriptions). Each set of results corresponds to a single pairwise comparison (e.g., cancer vs. normal) and will be named with the group names you assigned. Eight sets of results files are produced in the end:
- RNA-Seq or probe set-level - Feature-level statistics, exon annotations, AS/APS annotations, and functional predictions (protein, domain and miRNA binding site).
- Gene-level - Gene-level summary of data in feature-level file.
- Domain-level - Over-representation analysis of gene-level domain changes due alternative exon regulation.
- miRNA binding sites - Over-representation analysis of gene-level, predicted miRNA binding sites present in alternatively regulation exons.
- DomainGraph input file - Direct or inferred Affymetrix Exon 1.0 ST probe set IDs and summary statistics (see file #4) for analysis in DomainGraph. For non-exon arrays (e.g., RNA-Seq, Affymetrix junction or gene arrays), corresponding regulated exons are translated to the overlapping exon ID (see Analysis of AltAnalyze Results DomainGraph).
- All processed splicing scores - Feature-level statistics (see file #4) above for all analyzed features (not just significant). This is in the same format as the DomainGraph export file (see file #7).
- All feature-normalized intensities - (optional) Feature-level normalized intensities (see file #4) used to calculate splicing-index statistics or FIRMA fold changes for each sample. To obtain this file “Export all normalized intensities” option.
- Summary statistics file - Global statistics, reporting the number of genes alternatively regulated, number differentially expressed and summary protein association information (e.g., mean regulated protein length).
Each file is a tab delimited text file that can be opened, sorted and filtered in a spreadsheet program. These files are saved to the user-defined output directory under “AltResults/AlternativeOutput”, all with the same prefix (pairwise group comparisons). AltAnalyze will analyze all pairwise comparisons in succession and combine the feature-level and gene-level results into two additional separate files (named based on the splicing algorithm chosen).
Feature- and Gene-Level Alternative Exon Result Files
The feature-level file contains alternative exon data for either one probe set (exon-array), exon/junction IDs or reciprocal junctions (RNA-Seq and junction arrays). See Protein Direction Indicator for more information on these fields. In general, this file includes:
- Gene and feature annotations (e.g., description, symbol, exon/junction or probe set ID, feature exon ID, transcript clusters, links to Ensembl/UCSC exons, ordered exon-region IDs).
- Mean feature expression values for the regulated feature(s).
- Gene expression changes and baseline expression.
- Statistical results (e.g., splicing-index score, deviation value, normalized intensity p-value, adjusted normalized intensity p-value, MiDAS p-values, raw feature p-value).
- Alternative exon annotations (e.g., splicing-events, alternative promoters, alternative annotation confidence score).
- Protein- and microRNA-level associations (e.g., associated IDs, sequence, pattern of regulation, regulated domains/microRNA binding sites).
- Genomic coordinates of the regulated exon or pairs of reciprocal junctions regulated.
The gene-level file contains a summary of the data at the gene level, with each row representing a unique gene. This file also includes:
- Gene Ontology and pathway information for each gene obtained from Ensembl.
Protein Domain/Motif and miRNA Binding Site Over Representation Files
Over-representation analyses, (files 3 and 4) have the same structure:
- Column A is the name of the miR-BS or protein domain (e.g., sequence motif).
- Column B is the number of unique genes associated with alternatively regulated features for that sequence motif (aka Changed).
- Column C is the number of genes analyzed for over-representation that correspond to that sequence motif (aka Measured).
- Column D is the percentage Changed (Changed/Measured).
- Column E is the over-representation z-score (see Algorithms) for all unique genes aligning to the sequence motif that are alternatively regulated. A value of 1.96 is approximate to a p-value of 0.05 assuming a normal distribution.
- Column F is the Fisher Exact test p-value to assess the likelihood of this observation occurring by chance.
- Column G is the Benjamini-Hochberg adjusted p-value of F, to take into account multiple hypothesis correction.
- Column H contains all gene symbols for all unique genes changed.
Comparison Evidence File
A common question for biologists analyzing alternative exon profiles is which events are most likely true versus false positive predictions. RNA-Seq and junction microarrays allow for independent detection of alternative splicing events using only exon-junctions or only detected exons. The comparison evidence file examines results from the reciprocal junction analysis (ASPIRE or Linear Regression) and the single feature (exon or junction) analysis (splicing-index), to determine which events are predicted by both analyses or only be one. Those splicing-events predicted by both represented independently verified events that are most likely to represent valid known or novel alternative splicing events in the dataset. This file includes:
- Gene, exon and junction annotations (e.g., description, symbol, junction or probe set ID, feature exon ID, links to Ensembl/UCSC exons, ordered exon-region IDs).
- Statistical results (e.g., splicing-index score, deviation value, normalized intensity p-value, adjusted normalized intensity p-value, MiDAS p-values, raw feature p-value).
- Alternative exon annotations (e.g., splicing-events, alternative promoters, alternative annotation confidence score).
- Protein- and microRNA-level associations (e.g., associated IDs, sequence, pattern of regulation, regulated domains/microRNA binding sites).
- Algorithm from which the event was predicted (e.g., ASPIRE, splicing-index).
Diagnostic and Exploratory Visualization Results
In addition to the results files listed in the previous sections, various image plots are produced by AltAnalyze to assess quality control (QC), cluster gene or sample profiles (clustering) and identify associated cell types represented in each sample (Lineage Profiler). All plots are saved to the folder DataPlots in the user-defined output directory. Unlike the other AltAnalyze methods, these analyses require the installation of non-default Python packages if running directly from the Python source-code (see Installation). However, these dependencies are already included in the OS specific binary distributions.
Basic Quality Control
Multiple basic quality control plots are produced by AltAnalyze to evaluate sample quality and overall technical similarity to other samples in the dataset. Different QC metrics are applied based on whether the input data is from: (A) AltAnalyze normalized Affymetrix files, (B) RNA-Seq data or (C) pre-processed expression files.
If data from (A) applies, three output QC files will be generated: 1) distribution of normalized log2 probeset intensity values, 2) mean raw signal intensities of each array and 3) mean absolute deviation (MAD) of the RMA residuals for each array (Figure 2.18 A-C). The source data for all of these three QC metrics are derived from the Affymetrix Power Tools (see Pre-Processing, External Files and Applications) RMA analysis built into AltAnalyze[Lockstone2011].
If data from (B) applies, three groups of QC files will be generated: 1) distribution of log2 read-counts (exon and junction), 2) feature-level box-plots for the distribution of exon, junction and intron read-counts and 3) total number of reads for each sample, broken down by exon, junction and intron aligning (Figure 2.18 D-F). This source data for these plots is obtained from the file with the prefix counts in the folder ExpressionInput, which includes where each feature aligns to and the total number of associated read counts.
Data from (C) consist only of the distribution of log2 values in the input file.
Figure 2.18
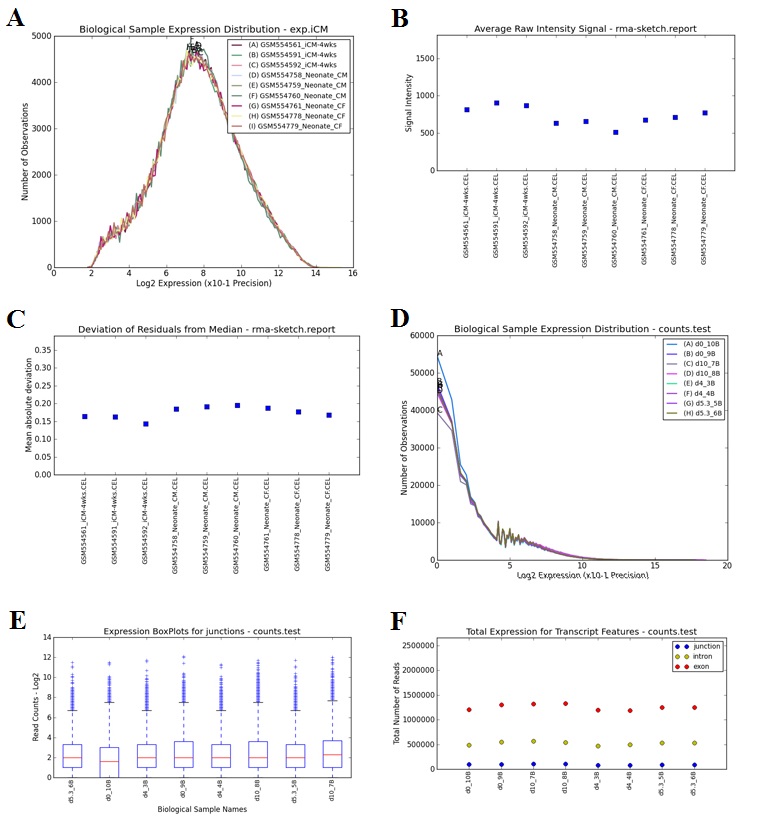
Basic QC Plots: Example AltAnalyze QC plots produced for normalized Affymetrix array (A-C) and RNA-Seq data (D-F). Only summarized junction count data is shown in E.
Expression Clustering
Two main expression clustering methods are currently output by AltAnalyze, hierarchical clustering and principal component analysis (PCA). Hierarchical clustering is used to identify overall patterns of gene expression shared by groups of genes and samples whereas PCA is used to visualize similarities between samples within and between groups in 2D dimension space. Hence, both methods can be used to evaluate the quality of the data as well as explore sample or gene relationships.
Figure 2.19
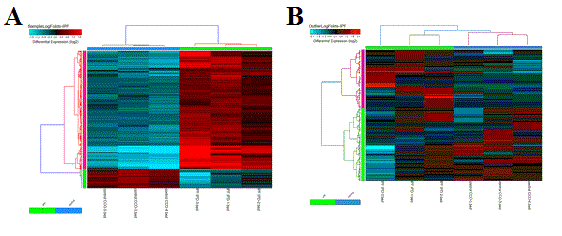
Hierarchical clustering heatmaps: Example AltAnalyze hierarchical clustering heatmaps for (A) significantly regulated genes in multiple comparisons and (B) outlier regulated genes. Genes are displayed as rows and samples as columns. Red indicates upregulation and blue indicates down. The vertical and horizontal bars adjacent to the heatmaps are colored based on a flat cluster threshold of 0.7 (distance criterion). In some cases, colors in the heatmap may not relate to colors in the dendrograms or flat cluster bars.
Hierarchical clustering is applied by default to both significantly differentially expressed genes and outlier regulated genes from the entire dataset. Genes considered significantly regulated are those that have greater than 2 fold (up or down) change in gene expression and comparison statistic p < 0.05 for any user indicated comparison (see GO-Elite options for details on changing the defaults - Figure 2.10). Outlier genes are those with a greater than 2 fold difference relative to the mean of all samples, for any gene not in the significantly regulated set. For significantly regulated genes, sample folds are calculated as compared to the mean of all samples for each gene or based on the group comparisons designated by the user (called “Relative”). Although default clustering metrics, methods and coloring options are applied to the resulting heatmaps, these options can be changed after running AltAnalyze (see Additional Analysis Options). Vector based versions of these plots are available in the PDF outputs in the folder DataPlots. A text file representing the clustered matrix, identifiers and flat-clusters will be exported to the DataPlots directory along with a TreeView compatible .cdt file. Note: only row names are included when the number of rows visualized is less than 100. Clustering is accomplished using Scipy’s cluster.hierarchy method. For additional details and workflows, see Tutorials.
PCA is applied only to the significantly differentially regulated gene set, by default. In this plot, the values of first component are plotted against the values of the second component for each sample (Figure 2.16). This analysis will visualize each sample as a colored circle, with the color corresponding to the different assigned biological groups. The sample names will be displayed to the right of the sample circle.
Lineage Profiler Analysis
Lineage Profiler is a new method introduced in AltAnalyze version 2.07. This algorithm correlates user supplied sample expression profiles with previously collected expression profiles from a large compendium of publicly available cell types and tissues (aka lineages). The underlying lineage data is biased towards adult, fetal and progenitor cell types arising throughout differentiation, as opposed to disease states or cell lines. It is capable of characterizing both microarray and RNA-Seq datasets for a diverse database of cell lineages.
The compendium itself is built on top of either exon or 3’array publically available datasets (human and mouse) combined from a large number of studies. From the entire compendium dataset, only the top 60 cell-specific markers are used for the lineage correlation analysis (see file AltDatabase/EnsMart65/uclidea/Hs/Hs_exon_tissue-specific_protein_coding.txt in the AltDatabase folder). The top markers are selected during the LineageProfiler database build process, based on their overall expression correlation to a specific-cell type relative to all other cell types examined. See Lineage Profiler for additional details on the algorithm.
Three main output files are currently provided by LineageProfiler: (1) sample-to-cell type correlation statistic flat-files, (2) hierarchically clustered heatmap of correlation statistics and (3) visualization of the correlation statics along a comprehensive lineage network. The primary statistic used form these analyses is a Z score calculated from the distribution of Pearson correlation coefficients for each user supplied RNA-profile to all analyzed lineages.
The correlation statistics flat-files are produced by LineageProfiler: (A) Pearson-correlation coefficients (ExpressionOutput/LineageCorrelations-\<dataset_name>.txt), (B) derived Z scores (ExpressionOutput/LineageCorrelations-\<dataset_name>-zscores.txt) and (C) average Z scores for each biological group (ExpressionOutput/Clustering/LineageCorrelations-\<dataset_name>-zscores-groups.txt). These files are all tab-delimited text files that can be easily explored in a spreadsheet viewer, such as Excel.
The hierarchically clustered heatmap output (2) is based on file (B). An example of this output is shown in Figure 2.19 B. This output is particularly useful for identifying changes in lineage associations during developmental transitions.
To further understand which cell fate decisions or lineage pathways are regulated in particular biological conditions, visualization along a comprehensive lineage network is provided (3) derived from file (C). An example of this output is shown in Figure 2.19 A. The lineage network is a community-curated network posted at WikiPathways. This network is visualized using the WikiPathways API (Wikipathways_webservice.py > viewLineageProfilerResults()). When running AltAnalyze from source-code, this function requires installation of the lxml library.
Additional à la carte Analyses
In addition to the streamlined AltAnalyze pipeline analyses, a number of individual useful functions can be run independently of these workflows. These include:
- Pathway Enrichment
- Pathway Visualization
- Hierarchical Clustering
- Principal Component Analysis
- Lineage Analysis and Sample Classification
- Network Analysis and Visualization
- Biological Identifier Translation
- Alternative Exon Visualization
- Venn Diagram Analysis
- File Merging Functionality
These functions provide a wide array of solutions for genomics analysis that are easily accessible to bioinformaticians and experimental biologists alike. These functions can be accessed through the Additional Analyses menu after selecting a species and platform type in the main menu. Alternatively, the functions can be run from the command-line for batch customized analytical and batch pipelines (see Running AltAnalyze from the Command-Line).
Pathway Analysis and Visualization
Pathway enrichment and visualization methods are identical to those provided in the independent analysis package GO-Elite. Enrichment analysis is available using a multiple algorithms, user defined thresholds and can be run on over a dozen distinct biological gene and metabolite categories. This tool provides an optimized list of enriched biological categories (e.g., Ontology term pruning) for description of input ID lists. In addition to tabular result files, hierarchically clustered heatmaps are displayed showing enrichment of terms between distinct conditions analyzed as well as networks of enriched terms with corresponding regulated genes. For more details see http://genmapp.org/go_elite/help_main.htm.
Hierarchical Clustering and Visualization
AltAnalyze can perform hierarchical clustering using default options (see Expression Clustering) or using customized options. These include the ability to change visualization modes (e.g., colors, contrast), clustering algorithm (e.g., cosine, uclidean, hopach), row normalization, matrix transposition and biological group coloring. In addition, several advanced options are available including the ability to cluster and visualize genes associated with certain GO-Elite pathways, ontologies or gene-sets and obtaining clusters of genes most correlated with a single candidate. These advanced options allow any users to easily and quickly obtain highly specialized expression views using a large selection set of biological categories, visualization options and advanced clustering algorithms from a single interface (Figure 2.20 A). More details on these options and parameters are described in Hierarchical Clustering Heatmaps.
Figure 2.20
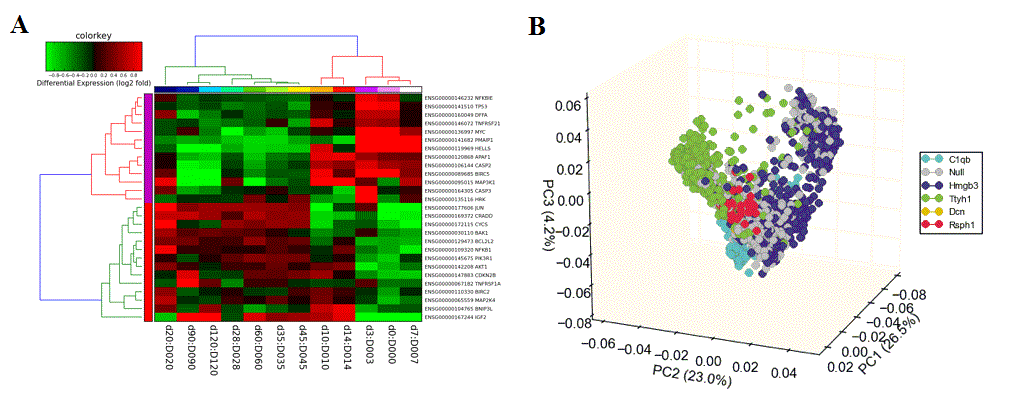
Advanced Clustering Options: Additional Analysis menu options available for hierarchical clustering of genes belonging to particular biological class (A) and PCA of samples in three dimensions (B), is shown. (A) Genes corresponding to the WikiPathway Apoptosis is displayed using the red-black-green color pallet and samples colored by biological category in the top color bar. (B) 3D PCA of Drop-Seq single-cell RNA-Seq for ICGS delineated genes with display of sample labels turned off. Cells are colored not by groups but by their relative expression for 5 ICGS reported guide-genes.
Principal Component Analysis (PCA)
In addition to the default pipeline output of two-dimension PCA plots (first two components), PCA can be run on its own using multiple customized options. These include optionally displaying sample labels and viewing a PCA plot interactively in three-dimensions as shown in Figure 2.20 B. The percentage of variance explained for each component is annotated in the component label. The top correlated and anti-correlated genes associated with the top four principal components are stored in the folder DataPlots/PCA.txt and can optionally be stored as an available gene set for other downstream analyses by entering a name for the analysis in the GUI.
Lineage Analysis and Sample Classification
This menu provides a number of flexible options for classifying samples relative to either (A) pre-compiled tissue/cell type references built from various transcriptome measurement platforms or (B) relative to a user supplied set of reference measurements. For tissue and cell-type classification, the LineageProfiler algorithm is employed, in which each loaded sample is matched to a set of tissue-specific markers determined from AltAnalyze’s MarkerFinder algorithm and then correlated to all available compendium cell type or tissue expression values. Although the overall correlation (Pearson correlation coefficient) between distinct platforms may be low (e.g., RNA-Seq versus exon array profiles), these sample specific correlations most typically are accurate, especially where many distinct sample types are being compared (manuscript in preparation). Results are output as a lineage correlation heatmap and as a WikiPathway network for lineage differentiation (DataPlots folder). The results in these files are z-scores derived from the distribution of observed correlations for all samples analyzed in a given experiment.
In addition to lineage classification, this algorithm can be applied to custom references and even distinct gene-models, discovered using the LineageProfilerIterate.py script provided with AltAnalyze. Using this method, samples can be classified using user-supplied references for all analyzed genes or subsets of gene-models provided in a gene-model file. Additional information on these methods, example workflows and example files can be found in Lineage Profiler and Sample Classification.
Network Analysis and Visualization (NetPerspective)
NetPerspective is a new tool introduced in AltAnalyze 2.0.8 that allows users to quickly and easily identify hypothetical biological networks between interacting genes, proteins, RNAs and metabolites with a single query. NetPerspective uses a collection of highly curated interactions from WikiPathways, KEGG and HMDB, experimentally derived transcription factor targets, annotated drug-protein interactions, microRNA target predictions (see Gene Annotation Assignment) and speculative protein interactions from BioGRID. Networks can be generated from lists of input IDs, existing interactions or GO-Elite pathways/gene-sets/ontologies, visualized with regulated gene, proteins and metabolites. Connections between sets of IDs can be identified using direct interactions, indirect or from the shortest path of possible connections. These networks are automatically displayed when run from the GUI and are also saved as PDF and PNG files to the folder network in the input file directory (Figure 2.21). When run from the command-line, automated generation of networks and images can be performed for an unlimited number of input lists run sequentially or in parallel. More details on these options and parameters are described in NetPerspective.
Figure 2.21
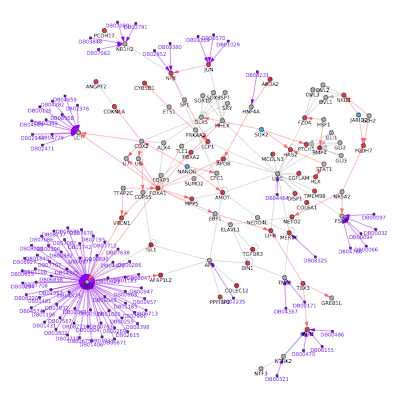
Automated Network Analysis and Visualization: Example output from NetPerspective. Nodes are colored as up or down-regulated (red or blue), with red edges indicating transcriptional regulation, blue indicating annotated inhibitory interactions, arrows indicating annotated directed interactions, purple arrows indicating drug interactions and green edges indicating microRNA-mediated interactions (not shown).
Alternative Exon Visualization (AltExonViewer)
An important means for initial validation of alternative exon expression (e.g., alternative splicing, alternative promoter regulation) is visualization of feature expression in the context of all measured gene features. In addition to visualization of alternative exons in the Cytoscape plugin DomainGraph (see Analysis of AltAnalyze Results DomainGraph), AltAnalyze contains a built in alternative exon viewer called AltExonViewer. This function allows users to display gene data in the form of a 1) line graph depicting exons along the X-axis and exon-expression or splicing index fold change along the Y-axis, 2) a heatmap of all exons across all samples and 3) a Sashimi-Plot genomic view. For the line graph option, expression values from each group are summarized as a single line color, with standard-error values included. One gene or multiple genes can be displayed at a time using a manual text entry field (e.g., SOX2 NANOG POU5F1 TCF7L1) or through a file selection option. Probed UTR regions and Introns can also be optionally displayed. To visualize exon-expression, select the raw expression option. This option requires that input expression files have already been generated and analyzed with AltAnalyze (conforming to the standard file locations - e.g., ExpressionInput/exp. File). To visualize alternative exon-expression directly, select the splicing-index option. This option works for already produced alternative exon results, which are saved to the folder AltResults/RawSpliceData (Figure 2.22). When analyzing a dataset with more than two groups, re-run the AltAnalyze workflow beginning with the Process AltAnalyze Filtered option and selecting the all groups selection for Comparisons to Perform option.
For the heatmap view, a standard AltAnalyze heatmap is produced with all exon region expression values (median normalized), ordered from beginning to end along the y-axis. The Sashimi-Plot option directly interfaces with the Sashimi-Plot source python code to produce high resolution splicing plots. Additional details on these options and parameters are described in Alternative Exon Visualization.
Figure 2.22
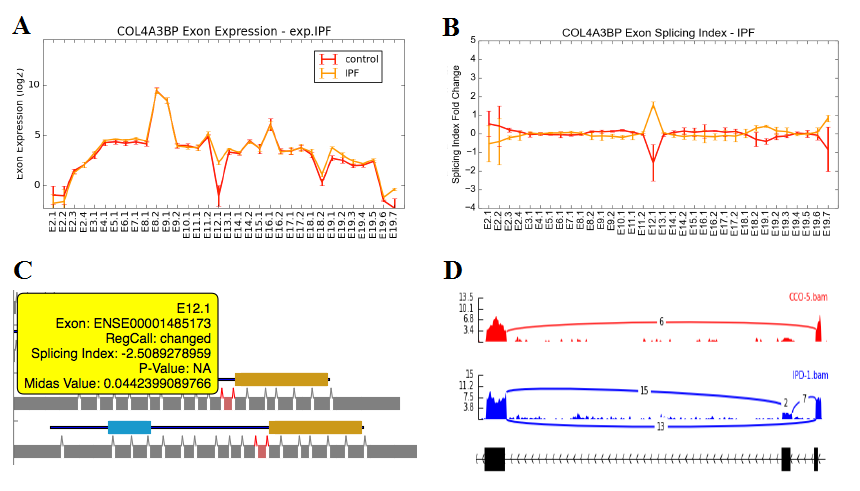
Alternative Splicing Visualization: Various techniques exist in AltAnalyze to visually confirm and evaluate alternative splicing. Results are shown for an example gene COL4A3BP for splicing of exon 12 (AltAnalyze database annotation). (A) In the first plot, three conditions two conditions are shown indicating the relative expression of exons corresponding to the below indicated exon regions (see Alternative Splicing Prediction). This view can be initiated from the AltAnalyze Additional Analyses > AltExon Viewer menu or the AltAnalyzeViewer software by right clicking on a gene name a selecting Exon Plot. (B) Analysis of a single pairwise-comparison of the two evaluated groups for exon-level splicing-index associated expression values. In both plots, E12.1 would be predicted to be “spliced-in” in the disease samples (also available from the AltExon Viewer). (C) Isoform viewer (aka SubgeneViewer) function available for visualizing protein (skinny black line) and domain encoding (blue and yellow blocks) regions corresponding to alternatively regulated exons and junctions (colored red for upregulation in the disease samples. This view can be initiated from the AltAnalyzeViewer accessory application, when right-clicking on a gene in any Tabular view and selecting Isoform Plot. The yellow box is initiated when mousing over an exon or domain feature to view annotations and/or associated statistics. (D) SashimiPlot visualization the specific detected splicing event (COL4A3BP) from the Sashimi Plot view in the AltAnalyzeViewer. Also available from the AltExon Viewer under Additional Analyses.
Venn Identifier Comparison Analyses
To evaluate commonalities and difference between different gene sets or other IDs obtained from AltAnalyze or outside programs, two tools are available within AltAnalyze for merging files and/or visualizing ID overlaps. To visualize the overlap between identifiers in two or more files (max of 4), select the Venn Diagram option in AltAnalyze (Additional Analyses menu). For this analysis, species and platform selection are not important. Select the different files of interest, containing comparable IDs in the first column of those files. Select an output directory for which you want the two types of Venn Diagrams to be saved to. Two methods are available for visualization of these diagrams: (A) Standard overlapping Venn’s and (B) ID membership weighted. The standard overlapping Venn will have equally sized circles or ovals representing IDs from each individual files (Figure 2.23). Selection of the associated numbers will prompt a new window to appear with the associated identifiers for that subset (automatically copied to your computers clipboard). The repository for the standard Venn diagram open-source code is no longer maintained, while the ID membership weighted Venn was obtained from the open-source project matplotlib-venn. This output will weight the circles in the Venn based on the relative overlap of IDs in each file (max of 3 files). Both of these outputs are automatically produced and saved to the indicated output directory with a time-stamp in the filename.
Figure 2.23
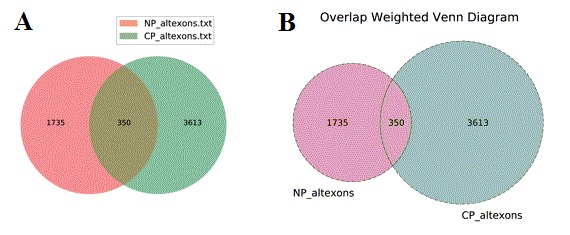
Venn Diagram Analysis in AltAnalyze: Venn Diagrams exported for two comparisons (alternative exons in neural and cardiac differentiation) for the (A) standard and (B) overlapping output image files.
File Merging Tool
Like the Venn diagram tool, this tool identifies differential overlaps between input identifier files and outputs a tab-delimited text file containing the original file contents for intersecting (Intersection option) or all combined IDs (Union). For this analysis, species and platform selection are not important. Up-to-four files can be selected for overlap. An output directory must also be selected for which to save the combined output (MergedFiles.txt) to. All columns contained in the original files will also be in the output with the column names followed by the source file (column-name.source-file.txt). Additional options are available for only returning unique IDs for each file or all possible combinations of matching IDs in the output (important when more than identical ID is present in the first column of a file).
Identifier Translation
A common use case for biologists dealing with genomics datasets is conversion of one identifier type to another. To accomplish this, users can access the Identifier Translation menu, load a file containing the IDs to be translated (must be the first column of values and obtain a new file in which the first column of values matches the desired ID type. These translations are accomplished through use of relationships obtained from Ensembl and HMDB (GO-Elite database > AltDatabase/EnsMart72/goelite/Hs/uid-gene). All original IDs and other column data will be present in the output file, along with the Ensembl or HMDB IDs used for translation. Where multiple Ensembl or HMDB IDs are related to the input ID, only one will be chosen (last listed).
AltAnalyze Results Viewer
The AltAnalyze Results Viewer is accessible through its own executable (AltAnalyze program directory), called the AltAnalyzeViewer or through the AltAnalyze Main menu (Figure 2.4, Interactive Result Viewer option). This viewer addresses a major challenge produced from the automated production of a massive archive of analysis results. As an example, if a user selects the option to perform pathway visualization, hundreds of colored WikiPathways image maps may be stored in multiple places within the output directory in a non-intuitive location. The AltAnalyze Results Viewer addresses this challenge by serving up the primary results in a more intuitive fashion that allows for fast navigation, access to data tables associated with each graphical views and interactive manipulation and creation of new graphical outputs. The interactive outputs include heatmaps with options to filter based on selected genes or pathways, gene-set enrichments within the heatmaps, PCA, interactive gene expression plots (DATASET file table view), SashimiPlots, Isoform Splicing Domain Plots and more (Figure 2.12). An example use of the viewer is shown here.