Multiple algorithms are available in AltAnalyze to identify differential alternative exon usage and alternative splicing. These algorithms that are compatible with exon-sensitive platforms and exon-exon junction sensitive platforms . For RNA-Seq we highly recommend using our algorithm MultiPath-PSI, which we have found has improved accuracy over other Percent-Spliced-In (PSI) approaches. Other splicing algorithms in AltAnalyze include splicing index method (exon or junction analysis), FIRMA (exon or junction analysis), MiDAS (exon or junction analysis), ASPIRE (junction analyses) and Linear Regression (junction analyses). For junction sensitive platforms (RNA-Seq and junction arrays), in addition to MultiPath-PSI, single feature analyses (e.g., splicing-index) are performed immediately after reciprocal junction analyses (e.g., ASPIRE) on the same list of expressed features. This allows the user to examine alternative exons predicted by pairs of reciprocal junctions in addition to those predicted by a single regulated exons/junctions (JP). In addition to these statistical methods, several novel methods are used to predict which alternative proteins correspond to a regulated exon, which protein domain/features differ between these and which RNA regulatory sequences differ between the associated transcripts (e.g., miR-BS).
Default Methods
The default options are stored in external text files in the folder “Config” as “defaults-expr.txt”, “defaults-alt_exon.txt”, and “defaults-funct.txt”.
| defaults-expr.txt | Default expression analysis options (Figure 2.6) |
| defaults-alt_exon.txt | Default alternative exon analysis options (Figure 2.7) |
| defaults-funct.txt | Default functional analysis options (Figure 2.7) |
These options correspond to those found in the configuration file “options.txt”. The user is welcome to modify the defaults and theoretically even the options in the “options.txt” file, however, care is required to ensure that these options are supported the by the program. Since AltAnalyze is an open-source program, it is feasible for the user to add new species and array support or to do so with AltAnalyze support. The default algorithms for the currently supported arrays are as follows:
| Exon/Gene | splicing-index (score > 2 and t-test p<0.05), no MiDAS |
| Junction/RNA-Seq | ASPIRE (score > 0.2 and permute p<0.05) |
| 3’ array | NA |
Algorithm Descriptions
Expression Normalization
AltAnalyze supports the comprehensive analysis of bulk and single-cell RNA-Seq, conventional and splicing microarray datasets. For all these, the software can quantify gene expression and evaluate differential gene expression. Different gene quantification methods exist depending on the types of files analyzed. For example, for FASTQ files, Kallisto analysis in AltAnalyze produces transcript and gene transcript-per-million (TPM) estimates, for BAM files RPKM values are calculated from aligned exon or junction reads and for microarrays, exon-associated probes are used to calculate gene normalized intensity values.
RNA-Seq FASTQ Files (TPM)
The direct analysis of FASTQ files is supported in AltAnalyze through an interface to the cross-platform compiled software Kallisto. Users downloading AltAnalyze for specific operating systems or from the source-code (GitHub) already have all Kallisto dependencies present. Kallisto provides pseudo-alignment of the RNA-Seq reads to estimate read counts per unique transcript (Ensembl gene model used in AltAnalyze) and transcript-per-million (TPM) estimates. TPM values for each gene are summed for each analyzed sample to obtain gene-level TPM values which are saved to the folder ExpressionInput in the expression file (prefix exp.). More information can be found on the Kallisto website.
RNA-Seq BAM Files (RPKM)
BAM files obtained from genome alignment software can be directly read into AltAnalyze for gene expression and splicing analysis. Exon and/or junction reads are determined from the BAM files and saved to .bed files for local analysis. These BED files contain junction and exon counts per region (junction or defined exon region). These counts can be analyzed directly in AltAnalyze, since alternative exon and junction expression are evaluated on a sample-by-sample basis, which allows for normalization of these values within a biological group and between group comparisons. However, gene expression will still be non-normalized when counts are used directly, hence, gene expression estimates may be incorrect, impacting all AltAnalyze gene expression fold changes. To account for this, the user can choose to normalize using the RPKM method. Users can choose between these normalization methods or no normalization methods (direct analysis of counts). Upon import of RNA-Seq counts, counts are incremented by 1, in order to calculate summary statistics without encountering zero-division errors. While the reported counts for gene expression and alternative splicing analyses will reflect the non-adjusted counts (e.g., zero, where applicable) for raw and quantile normalized counts, RPKM values will always reflect the adjusted values. In cases where RPKM adjusted counts are selected, an original zero read count in two compared groups will be interpreted as a fold change of 1, rather than reflecting the actual adjusted RPKM comparison value (values will differ between samples since the total number of reads per sample will vary).
Affymetrix Microarrays
For Affymetrix array analyses, AltAnalyze calls the software Affymetrix PowerTools, distributed with the GPU license. For conventional 3’ expression arrays, the RMA methods is applied to all Affymetrix CEL files (all supported formats) in the input user directory. Expression values are computed based on corresponding probeset annotations in the downloaded CDF file (automatically recognized and downloaded by AltAnalyze or supplied by the user). For exon, gene and junction arrays, RMA is also applied but for exon/junction-level probesets and not transcript cluster probesets (gene-level). In addition, detection above background (DABG) p-values are calculated for these arrays to evaluate likelihood of exon expression for calculating gene expression estimates and assessing probeset-level alternative exon expression (see Probe Set Filtering and Constitutive and Gene Expression Calculation).
Agilent Microarrays
Agilent Feature Extraction files can also be loaded and normalized using a workflow similar to that for Affymetrix microarrays. Agilent Feature Extraction files are produced from Agilent scanned slide images using Agilent’s proprietary Feature Extraction Software. Feature Extraction text files can be loaded in AltAnalyze using the Process Feature Extraction Files option and selecting the appropriate color ratio or specific color channel from which to extract expression values from. Quantile normalization is applied by to Agilent data processed through this workflow.
For all additional expression dataset types, both quantile normalization and batch-effects removal are available. Batch effect removal is provided using the combat python package, which provides nearly identical results to that of the R version of combat. See Batch Effect Correction in AltAnalyze Using Combat for additional information. In addition, to adjust for differences between distinct biospecimens where paired measurements are available, such as a treatment time-course or regional tissue dissections, you can select the option groups under Normalize feature expression in the Expression Dataset Parameters window. Similar to the batch effects analysis, the user will be able to indicate which samples correspond to the same biospecimen. Subsequently, AltAnalyze will calculate the mean expression of each gene for the same biospecimen samples and then normalize the expression of each value relative to that biospecimen specific mean. As a result, the t-test p-values will break down to a paired t-test p-value and biospecimen specific effects will be removed. An example is described in the following publication[Singer2015].
Gene Expression Analysis
For the simple gene expression output files saved to the ExpressionOutput directory, several basic expression statistics are calculated. These statistics are performed for any user specified pairwise comparisons (e.g., cancer versus normal) and between all groups in the user dataset (e.g., time-points of differentiation). Expression values are reported as log2 values for all microarray analyses and as non-log values for RNA-Seq analyses. These statistics are comprised of the following: (1) rawp, (2) adjp, (3) log-fold, (4) fold change, (5) ANOVA rawp, (6) ANOVA adjp and (7) max log-fold. The rawp is a one-way analysis of variance (ANOVA) p-value calculated for each pairwise comparison (two groups only). The adjp is the Benjamini-Hochberg (BH) 1995 adjusted value of the rawp. The log-fold is the log2 fold calculated by geometric subtraction of the experimental from the control groups for each pairwise comparison. The fold change is the non-log2 transformed fold value. The ANOVA rawp is the same as the comparison rawp, but for all groups analyzed (note, this is reduced to the rawp when only two groups are analyzed). The ANOVA adj is the BH adjusted of the ANOVA rawp. The max log-fold is the log2 fold value between the lowest group mean and the highest group expression mean for all conditions in the dataset. These statistics are intended for further data filtering and prioritization in order to assess putative transcription differences between genes. For non-RPKM RNA-Seq analyses, these values will be the average of exon or junction read counts (exons when both present) for known or constitutive annotated features. When RPKM normalization is selected, gene expression will be based on the RPKM of all “expressed” exons or junctions in any of the sample groups (user defined filtering thresholds), taking into account the total read counts for “expressed” exons, their sequence length and the total number of reads per sample. Filtering thresholds for exons and junctions are separate variables, since the likelihood of obtaining a junction read count are typically lower than an exon read count, due smaller sequence regions and algorithms available to identify both. To calculate a fold-change, the final gene-level counts are incremented by 1, prior to calculating the RPKM. These same gene RPKM values are used for alternative exon analyses. An additional column is present for RNA-Seq analyses with the total reads per gene shown, in order to filter for low versus high absolute expression (also available from the file “counts.YourDataset-steady-state.txt”).
Statistical Testing Procedures
Multiple conventional statistical tests are provided for the various comparison analyses available (e.g., differential gene expression, alternative exon expression). For group comparisons involving greater than two groups, a standard f-test statistic is used. For pairwise group comparisons, users can choose from unpaired and paired t-test’s (assuming equal variance), rank sum, Mann-Whitney and Kolmogorov-Smirnov. These tests are made available from the open-source statistical package SalStat. In addition to these un-moderated tests, AltAnalyze provides a moderated t-test option (unpaired, assuming equal variance), based on the limma empirical Bayes model. This model calculates an adjusted variance based on the variance of all genes or exons analyzed for each comparison. Unlike a standard limma analysis, only the variances for the two groups being compared are used to compute the adjusted variance (s2 prior and df prior) for each comparison as opposed to variance from all groups analyzed (when greater than 2). For these calculations (gamma functions), AltAnalyze uses the python mpmath library. For q-value calculations, AltAnalyze imports the python adapted qvalue library.
Single-Cell Heterogeneity Analysis with ICGS
As an unsupervised approach to identify gene- and cell-population heterogeneity within large single-cell and bulk RNA-Seq datasets, a novel algorithm called ICGS has been developed for AltAnalyze. Iterative Clustering and Guide-gene Selection (ICGS) utilizes pair-wise correlation of dynamically expressed genes and iterative clustering with pattern-specific guide genes to delineate coherent gene expression patterns. This algorithm employs an iterative filtering, correlation, clustering and guide selection workflow that results in a coherent set of differentially expressed genes and predicted sample groups. By itself, these gene sets can be utilized for further investigation (e.g., lineage differentiation pathways). Both the ICGS menu option and the hierarchical clustering Additional Analyses menu can perform the major elements of this analysis. (Step 1) The algorithm begins by importing all supplied expression values from an input dataset, empirically determining whether the data should be log2 adjusted, filtering the data based on the supplied RPKM and maximal gene count expression thresholds, examining only protein coding genes in the AltAnalyze Ensembl gene database and searching for genes with at least N number of samples differing between for a given gene based on the indicated fold cutoff (e.g., the third lowest expressing gene versus the third highest expression gene, with at least a 4 fold difference, for n=4 and a minimum 4 fold difference). (Step 2) This results in a filtered gene set from which to perform all pairwise correlations (Pearson correlation > 0.4, by default) and select only those genes that are correlated to at least 10 distinct genes. Because an individual sample can guide the correlations, only gene sets that remain intra-correlated following removal of the most highly expressed gene are retained (except in cases where only one sample difference are selected from the Minimum number of samples differing option). This pioneering round gene set is clustered using the supplied clustering algorithms (cluster 1) and used for further downstream filtering. (Step 3) In the next round, iterative filtering steps are applied using different correlation thresholds to select no more than 20 genes for each clustered “pattern”, which are then re-clustered (cluster 2). (Step 4) The resulting cluster is re-supplied to the prior algorithm, with additional option to select to select the top correlated gene(s) for each cluster (guides) to other genes in that gene cluster. Multiple transcription factor genes are selected from each cluster, if present; otherwise a single guide-gene is selected. If the user indicated that cell-cycle genes should be excluded, any clusters from this set with more than one cell-cycle gene will be excluded. These guides genes are then correlated to those produced from Step 1 (filtered from the original set based on expression, annotation and fold change) using the user supplied correlation cutoff to identify positively correlated genes (maximum of 50 per guide gene) (cluster 3). (Step 5) This last step is re-iterated using the results of cluster 3 to further refine the results (cluster 4). All clustered options are presented to the user for sample groups selection, for downstream AltAnalyze workflow differential expression and splicing analyses (if applicable). When performing this workflow for alternative exons (AltExon option), only the Minimum number of samples differing is considered and is applied to the AltAnalyze MultiPath-PSI in analysis (see algorithm details below).
Reciprocal Junction Analysis
Alternative exon analyses differ for exon-based analyses as compared to junction sensitive analyses. For an exon-level analysis, alternative exon regulation is determined by comparing the normalized expression of one exon in two or more conditions (pairwise versus all groups analysis). Normalized expression is the expression of an individual exon (aka a probe set) divided by the gene-expression value (collective expression of gene features that best indicate constitutive versus alternative expression). In contrast, junction sensitive analyses (e.g., RNA-Seq and junction arrays) assess the expression of junctions often in addition to exons. For alternative splicing and often alternative promoter selection, two junctions (or an inclusion exon and exclusion junction) are differentially expressed in opposite directions, leading to the inclusion or exclusion of exons, exon-regions or introns. Thus, these methods are more sensitive in detecting alternative exon expression and the precise splicing-event that is occurring, as opposed to identifying on the exon features that are regulated (Figure 3.1). Although this method is more accurate at detecting true alternative splicing events, single exon or junction analyses are still useful at detecting other forms of alternative exon-region expression (e.g., alternative promoter selection and alternative 3’ end-processing). While junction arrays contain exon features for assessing alternative 3’ end-processing, RNA-Seq junction-only data does not, thus, details on alternative polyadenylation will likely be missing.
Figure 3.1
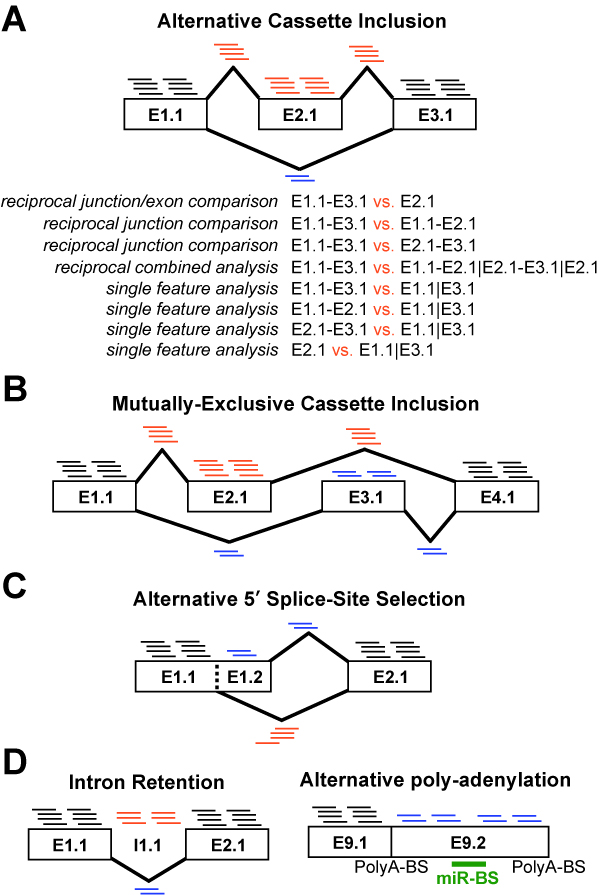
Splicing and Alternative Exon Events Detected by Different Algorithms: Examples are shown for features (RNA-Seq reads or Affymetrix probe sets) that can be used to detect distinct alternative events. A) Alternative cassette-exon inclusion is shown with increased inclusion of exon E2.1 (AltAnalyze exon block-region notation). Exon E2.1 and junctions E1.1-E2.1 and E2.1-E3.1 (inclusion junction) are expressed higher in this example (more associated reads) relative to junction E1.1-E3.1 (exclusion junction). While this indicates that alternative splicing is occurring in one condition, AltAnalyze looks at changes in the expression or ratio of expressed isoforms between at least two experimental conditions. Listed are the possible exon and junction analyses available to infer an alternative cassette splicing is occurring between two conditions. These include 1) comparison of the exclusion junction to the inclusion exon, 2) comparison of the exclusion junction and one of the inclusion junctions, 3) comparison of the exclusion junction and the mean expression of all inclusion features and 4) comparison of any exclusion or inclusion features relative to exons that are not alternatively spliced. Options 1-3 are performed only by junction arrays and exon/junction RNA-Seq using reciprocal junction analysis (e.g., ASPIRE), options 2 and 3 are currently performed by RNASeq, while option 4 is used for junction-arrays, exon arrays and RNASeq (e.g., splicing-index). Figures B-D represent other examples of alternative exon inclusions. None of the examples in Figure D can be detected by junction-only RNA-Seq analyses.
To identify alternative exons from junction analyses, AltAnalyze first identifies reciprocal junctions or pairs of exon-junctions, one measuring the inclusion of an exon and the other measuring exclusion of the exon. This algorithm allows for the identification of novel exon junctions detected by RNA-Seq that overlap in the genome with known junctions and splice to at least one known splice site. If exon expression is also measured, an exclusion junction can be directly compared to the expression of an included exon. For the reciprocal junction algorithms used by AltAnalyze (ASPIRE, Linear Regression, Ri-PSI), the expression of the exclusion junction is directly compared to the expression of the inclusion junction or exon for each sample. In this way, these algorithms don’t require normalization of expression, which is useful for RNA-Seq analyses were read counts are directly used. ASPIRE does use the gene expression estimate as an additional factor, however, since these values are calculated on a per sample basis, normalization is not an issue. Pairs of reciprocal junctions or reciprocal junction/exon pairs are extracted from the AltAnalyze gene database for known junctions (Ensembl and UCSC) by comparing the exon block and region structure of transcripts (see Building Ensembl-Exon Associations) in addition to at runtime for novel junctions detected from an RNA-Seq experiment. For junction arrays, novel reciprocal junctions are included in the gene database from Affymetrix predicted junctions, using the same exon block and region comparison strategy. For the AltMouse array, reciprocal probe set pairs are determined using the ExonAnnotate_module.py module using the “identifyPutativeSpliceEvents” function, by comparing AltMerge exon block and region annotations from Affymetrix. These methods allow for the identification of cassette exon inclusion, alternative 5’ and 3’ splice-site selection, mutually-exclusive cassette inclusion, alternative promoter selection, intron-retention and trans-splicing (RNA-Seq only).
Splicing Index Method
This algorithm is described in detail in a number of publications[Srinivasan2005][Gardina2006]. Briefly, the expression value of each RNA-Seq feature or probe set for a condition is converted to log space (if necessary). For each feature examined, its expression (log2) is subtracted from the mean gene expression of value to create a gene expression corrected log ratio (subtract instead of divide when these values are in log space). This value is the normalized intensity. This normalized intensity is calculated for each experimental sample, using only data from that sample. To derive the splicing-index value, the group-normalized intensity of the control is subtracted from the experimental. This value is the change in exon-inclusion (delta I, dI, or splicing index fold change).

An f-test p-value is calculated (two tailed, assuming unequal variance) by comparing the normalized intensity for all samples between the two experimental groups. A Benjamin-Hochberg adjusted p-value from this f-test p is also calculated. A negative SI score of -1 thus indicates a two-fold change in the expression of the RNA-Seq feature or probe set, relative to the mean gene expression, with expression being higher in the experimental versus the control. When more than two-groups are being compared in AltAnalyze (“all groups” or “both options" for “Type of group comparisons to perform”), the splicing-index value is calculated between the two biological groups with the lowest and highest normalized intensities. The two groups being compared are indicated in the alternative exon results file.
FIRMA Analysis
FIRMA or Finding Isoforms using Robust Multichip Analysis[Purdom2008] is an alternative to the splicing-index approach, to calculate alternative splicing statistics. Rather than using the probe set expression values to determine differences in the relative expression of an exon for two or more conditions, FIRMA uses the residual values produced by the RMA algorithm for each probe, corresponding to a gene. The median of the residuals for each probe set, for each array sample is compared to the median absolute deviation for all residuals and samples for the gene.
Although the core FIRMA methods are the same as the original implementation in R, AltAnalyze FIRMA differs in several important ways:
-
The standard AltAnalyze core, extended, or full probe set definitions define the probe composition of each gene, rather than the Affymetrix transcript cluster definitions. Thus, each probe must correspond to a single Ensembl gene to be analyzed.
-
While FIRMA scores for each sample and probe set are calculated, only summary statistics are reported in the standard AltAnalyze output files. This statistic is the average FIRMA score for all samples in the experimental group minus the average of the FIRMA scores in the designated baseline group. If no comparisons are specified, the two groups with the largest difference in scores are reported.
-
FIRMA scores are organized into groups for calculation of summary statistics (FIRMA fold change and t-test p-values). However, scores for each probe set and sample can be optionally exported.
Users can define whether to use the AltAnalyze core, extended or full annotations in the program interface or using the --probetype flag in the command-line interface. An unique numerical ID corresponding to each Ensembl gene and all associated gene probe sets for FIRMA analysis are stored in a metaprobeset file in the array annotation directory (e.g., AltDatabase/EnsMart54/Hs/exon/Hs_exon_core.mps). This file is used to define gene level probe sets by the program APT.
Similar to splicing-index analysis of exon-tiling data, expression and DAGB filtering of probe set expression, FIRMA score p-value filtering and gene expression reporting/filtering based on either constitutive or all exon aligning probe sets. To export FIRMA scores for each probe set and sample, select the option “Export all normalized intensities” from the “Alternative Exon Analysis Parameters” window, or by using the flag --exportnormexp in the command-line interface.
MiDAS
The MiDAS statistic is described in detail in this white paper. This analysis method is available from the computer program APT, mentioned previously. APT uses a series of text files to examine the expression values of each probe set compared to the expression of user supplied gene expression reporting probe sets. This method can also be used with RNA-Seq junctions when performing single feature-level analyses. Since AltAnalyze uses only features found to align to a single Ensembl gene (with the exception of RNA-Seq for trans-splicing events), AltAnalyze creates it’s own unique numerical gene identifiers (different than the Affymetrix transcript clusters). When written, a conversion file is also written that allows AltAnalyze to translate from this arbitrary numerical ID back to an Ensembl gene ID. These relationships are stored in the following files along with the feature expression values:
| meta-Hs_Exon_cancer_vs_normal.txt | Relates feature to gene |
| gene-Hs_Exon_cancer_vs_normal.txt | Gene expression values (non-log) |
| exon-Hs_Exon_cancer_vs_normal.txt | Feature expression values (non-log) |
| commands-Hs_Exon_cancer_vs_normal.txt | Contains user commands for APT |
| Celfiles-Hs_Exon_cancer_vs_normal.txt | Relates sample to group |
| probeset-conversion-Hs_Exon_cancer_vs_normal.txt | Relates arbitrary gene IDs back to Ensembl |
When the user selects the option “Calculate MiDAS p-values”, AltAnalyze first exports expression data for selected RNA-Seq feature (e.g., AltAnalyze “core” annotated - Figure 2.7) to these files for all pairwise comparisons. Once exported, AltAnalyze will communicate with the APT binary files packaged with AltAnalyze to run the analysis remotely. MiDAS will create a folder with the pairwise comparison dataset name and a file with MiDAS p-values that will be automatically read by AltAnalyze and used for statistical filtering (stored in the AltAnalyze program directory under “AltResults/MiDAS”). These statistics will be clearly labeled in the results file for each feature and used for filtering based on the user-defined p-value thresholds (Figure 2.7). When more than two-groups are being compared in AltAnalyze (“all groups” or “both options for “Type of group comparisons to perform”), the MiDAS p-value is reported for all groups designated by the user in combined array files or expression dataset file.
Note: Different versions of the APT MiDAS binary have been distributed. AltAnalyze is distributed with two versions that report slightly different p-values. The older version (1.4.0) tends to report larger p-values than the most recent distributed (1.10.1). Previous versions of AltAnalyze used version 1.4.0, while AltAnalyze version 1.1 uses MiDAS 1.10.1 that produces p-values that are typically equivalent to those as the AltAnalyze calculated splicing-index p-values (larger).
ASPIRE
For exon-exon sensitive junction platforms (RNA-Seq, HJAY, MJAY, AltMouse), the algorithm analysis of splicing by isoform reciprocity or ASPIRE was adapted from the original report[Ule2005] for inclusion into AltAnalyze. Similar to the splicing-index method, for each reciprocal junction, a ratio is calculated for expression of the junction (non-log) divided by the mean of all gene expression reporting junctions and exons (non-log), for the baseline and experimental groups. The ASPIRE dI is then calculated for the inclusion (ratio1) and exclusion (ratio2) junctions, as such:
Rin = baseline_ratio1/experimental_ratio1
Rex = baseline_ratio2/experimental_ratio2
I1 = baseline_ratio1/(baseline_ratio1+baseline_ratio2)
I2 = experimental_ratio1/(experimental_ratio1+experimental_ratio2)in1 = ((Rex-1.0)*Rin)/(Rex-Rin)
in2 = (Rex-1.0)/(Rex-Rin)
dI = -1*((in2-in1)+(I2-I1))/2.0
If (Rin>1 and Rex<1) or (Rin<1 and Rex>1) and the absolute dI score is greater than the user supplied threshold (default is 0.2), then the dI is retained for the next step in the analysis. By default, AltAnalyze will calculate a one-way ANOVA p-value for the sample ASPIRE scores (relative to the compared condition group means) and a false-discovery rate p-value of this one-way ANOVA (Benjamin-Hochberg). In place of these statics, the user can choose to perform a permutation analysis of the raw input data to determine the likelihood of each ASPIRE score occurring by chance alone. This permutation p-value is maximally informative when the number of experimental replicates is > 3. This permutation p-value is calculated by first storing all possible combinations of the two group comparisons. For example, if there are 4 samples (A-D) corresponding to the control group and 5 (E-H) samples in the experimental group, then all possible combinations of 4 and 5 samples would be stored (e.g, [B, C, G, H] and [A, D, E, F]). For each permutation set, ASPIRE scores were re-calculated and stored for all of these combinations. The permutation p-value is the number of times that the absolute value of a permutation ASPIRE score is greater than or equal to the absolute value of the original ASPIRE score (value = x) divided by the number of possible permutations that produced a valid ASPIRE score ((Rin>1 and Rex<1) or (Rin<1 and Rex>1)). If this p-value is less than user defined threshold, or x<2 (since some datasets have a small number of samples and thus little power for this analysis), the reciprocal junctions are reported.
Linear Regression
When working with the same type of reciprocal junction data as ASPIRE, a linear regression based approach can be used with similar results. This method is based on a previously described approach[Sugnet2006]. This algorithm uses the same input ASPIRE (junction comparisons, constitutive adjusted expression ratios). To derive the slope for each of the two biological conditions (control and experimental), the constitutive corrected expression of all samples for both reciprocal junctions is plotted against each other to calculate a slope for each of the two biological groups using the least squared method. In each case, the slope is forced through the origin of the graph (model = y ~ x - 1 as opposed to y ~ x). The final linear regression score is the log2 ratio of the slope of the baseline group divided by the experimental group. This ratio is analogous to a fold change, where 1 is equivalent to a 2-fold change. When establishing cut-offs, select 2 to designate a minimum 2-fold change. The same permutation analysis used for ASPIRE is also available for this algorithm.
MultiPath-PSI Splicing Algorithm
To enable fast and accurate analyses of both bulk and single-cell RNA-Seq, we adapted a recently developed Percent Spliced In (PSI) splicing method called MultiPath-PSI. This algorithm is introduced in AltAnalyze version 2.1.1, requiring aligned BAM files as input. Similar to other recent reported local splicing variation (LSV)-based methods, such as MAJIQ (https://majiq.biociphers.org) and LeafCutter (https://github.com/davidaknowles/leafcutter), MultiPath-PSI considers the detected junctions within a restricted genomic interval for assessing local junction expression differences. Unlike these alternative approaches, MultiPath-PSI considers all known and novel exon-exon junctions in a sample or cell and computes its relative detection compared to the local background of all genomic overlapping junctions that can be directly associated with the given gene of interest (sharing at least one known gene-associated splice-site) Figure 3.2.
This calculation provides a more inclusive and conservative estimate of LSV. These same junctions are used to identify high confidence intron retention splicing events evidenced by pairs of exon-intron and intron-only mapping paired-end reads, sufficiently detected at both ends of a given intron (5 and 3). This more stringent algorithm requires that only counts for exon-intron spanning reads are reported, in which multiple exon-intron spanning reads are detected at both ends of the intron and a matching paired-end read contained entirely within the intron is present (BAMtoExonBed module of AltAnalyze). PSI values are calculated for any junction with a minimum PSI difference of 0.1 (10% PSI) between any sample or cell. PSI values are only reported for samples or cells for a given splicing event in which sufficient read-depth is present (minimum of 20 reads per examined junction interval). Junctions are clustered into groups of unique junction clusters when reporting the results to identify redundant splicing events. Unique junction cluster IDs are determined by examining the overlap in exon-exon and exon-intron junctions for a given gene to identify connected subgraphs in the exon-exon/exon-intron network.
To ensure the approach is accurate, we benchmarked this approach against several recent reported methods for local splicing variation analyses (MAJIQ, LeafCutter, rMATS). A gold-standard to evaluate prediction accuracy and error rates, we developed a simulation RNA-Seq dataset derived from known and novel isoform predictions for pluripotent stem cells (PSC) and in vitro derived day 30 cardiomyocytes (CM). To create this simulation dataset, we first predicted known and novel isoforms and expression estimates using Cufflinks for biological triplicate samples (PSC and CM). The Cufflinks isoform GTF and expression values (isoforms.fpkm_tracking) were supplied as input for the software Polyester to produce simulated RNA-Seq reads at a depth of 100 million paired-end reads using the software default parameters 7. These subsequent FASTA files were supplied as input for genome alignment (hg19) with the software STAR to produce BAM files with read-strand predictions. The associated aligned sequencing data was evaluated with each analysis methods and benchmarked against isoform-derived alternative splicing events (exon-exon and exon-intron junctions) comparing CM and PSC. Supervised group comparison analyses with MultiPath-PSI are performed using an FDR corrected (Benjamini-Hocherg) empirical Bayes moderated t-test P-value < 0.05. For each method, a ranked list of splicing events were ordered according to the empirical score or p-value reported by each algorithm for all reported events. Precision and recall curves were generated in Matlab. Comparison of unique splice-junction clusters from each algorithm (determined from the junction-graph of junction genomic coordinates output from each method) to these gold-standards, indicated that MultiPath-PSI has the greatest overall accuracy and lowest error rates, based on precision and recall estimates. These same trends were reproduced when considering each specific primary mode of splicing regulation (cassette-exon, alternative splice-site, intron retention), with intron retention showing the greatest gains in precision and recall from MultiPath-PSI.
Raw MultiPath-PSI results are reported in the file: AltResults/AlternativeOutput/Hs_RNASeq_top_alt_junctions-PSI_EventAnnotation.txt. This file includes splicing functional predictions (e.g., splicing-event type, protein-level impacts), unique junction-cluster IDs and genomic positions of the corresponding junctions. From these results, the software will output moderated t-test p-values with and without Benjamini-Hochberg (rawp and adjp, respectively), filtering for an adjp < 0.05 and dPSI>0.1 to the folder AltResults/AlternativeOutput/Events-dPSI_0.1_adjp for all AltAnalyze indicated comparisons.
Figure 3.2
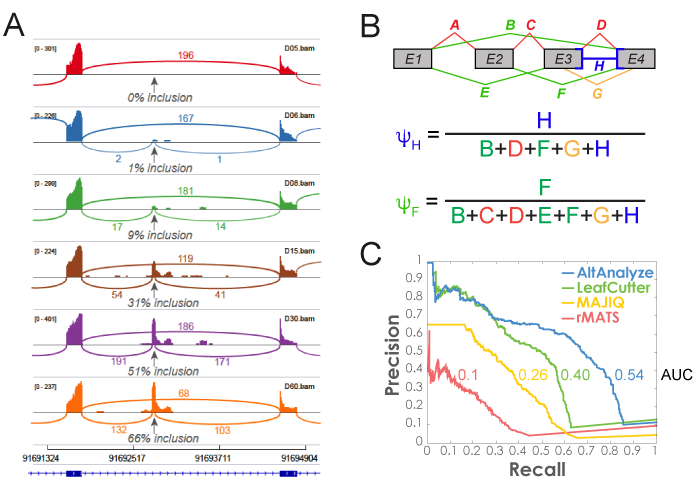
MultiPath-PSI Algorithm: Alternative splicing algorithm design (MultiPath-PSI) for single-cell and bulk RNA Seq analyses implemented in the software AltAnalyze. A) PSI example and B) example equations for calculating intron retention or alternative exon-exon junction Percent Spliced In (PSI) values from associated aligned paired-end read counts associated with genomic overlapping junctions (denominator) with an examined reference junction (numerator). Each letter indicates a respective exon-exon junction, for exon-inclusion splice junctions (red), exon-skipping junctions (green), alternative splice-site associated junctions (orange) or multi-evidence exon-intron retention junction counts (blue). C) Precision and recall curves for splicing event detection were calculated based on ranked algorithm confidence scores. Gold standard splicing events were derived from isoform expression estimates used to produce simulated (Polyester) RNA-seq reads.
External Alternative Exon Analysis File Import
In addition to providing several alternative exon analysis algorithms options in AltAnalyze (MiDAS, FIRMA, splicing-index), users can import Affymetrix alternative exon results from other programs for downstream functional annotation analyses. These analyses consist of alternative exon annotation (alternative splicing and alternative promoter selection), protein isoform, protein domain and microRNA binding site disruption analysis and pathway over-representation. Two options for external probe set analysis are now available:
-
Annotation of third party alternative exon data in AltAnalyze
-
Restricted analysis of probe sets using a pre-defined list
Both options are conceptually similar; provide AltAnalyze with a list of probe sets and optionally statistics, and AltAnalyze will return alternative exon statistics (option 2) and/or functional annotations for the input probe sets (option 1 and 2).
Option 1 - Import and annotation of third party probe set results
For this option, no raw data is required (e.g., CEL files or expression values) just a list of probe sets of interest. This option is simply available by selecting the main analysis option “Annotate External Results” and selecting a tab-delimited probe set list. The probe sets supplied in this list can be produced by any alternative exon analysis program the user prefers, as long as the output is exon-level Affymetrix probe sets. Users can provide direct output files from JETTA or provide results in a more generic format, consisting of regulated probe sets and result statistics (optional).
The generic file format for alternative exon results import is:
-
(Column 1) Probe set ID (required)
-
(Column 2) Alternative-exon fold (optional)
-
(Column 3) Alternative-exon p-value (optional)
-
(Column 4-100) Ignored data (optional)
In addition to this generic format, results from the program JETTA can also be directly imported. Since this method produces two p-values for the MADS algorithm, the smallest of the two p-values is used. The results from this analysis are typical AltAnalyze result files, including input for DomainGraph and include any alternative exon statistics supplied with the probe set list.
Option 2 - Restricted probe set analysis (Advanced Feature)
This option can be performed with any of the main AltAnalyze analysis options (e.g., Process CEL Files, Expression Files or AltAnalyze Filtered). It is triggered by supplying a list of probe sets for each comparison of interest in the directory “filtering” in the AltAnalyze program directory (AltAnalyze_v1release/AltDatabase/filtering). The restricted filtering files are tab-delimited text files containing the corresponding exon probe set ID (Column 1) and optionally exon statistics and annotations (Columns 1-100). While the statistics and annotations provided in the restricted filtering file are not used for any computation, they are appended as extra columns in the alternative exon results file. Multiple files, corresponding to specific biological comparisons (e.g., Tumor_vs_wild-type) can exist in this folder and only those files who’s name matches the comparison name will be matched and used for restricted analysis. For example, if biological sample groups A, B and C are being compared, to filter C_vs_A, the restricted probe set file must be named C_vs_A.txt. Note: If the user enters any filters (e.g., maximum DABG p value threshold, minimum alternative exon score), these will further restrict which “significant” probe sets are reported. To report all probe sets found in the AltAnalyze database, make sure to set all thresholds to the minimum or maximum value (e.g., --dabgp 1 --rawexp 1 --altp 1 --probetype full --altscore 1 --GEcutoff 10000 - see Running AltAnalyze from the Command-Line). Alternatively, users working with the command-line interface can use the option --returnAll to automatically enter this minimum values (see Running AltAnalyze from the Command-Line).
Domain/miR-BS Over-Representation Analysis
A z-score is calculated to assess over-representation of specific protein sequence motifs (e.g., domains) and miR-BS’s found to overlap with a probe set or RNA-Seq (feature) that are alternatively regulated according to the AltAnalyze user analysis. This z-score is calculated by subtracting the expected number of genes in with a protein feature or miR-BS meeting the criterion (alternatively regulated with the user supplied thresholds) from the observed number of genes and dividing by the standard deviation of the observed number of genes. This z-score is a normal approximation to the hypergeometric distribution. This equation is expressed as:
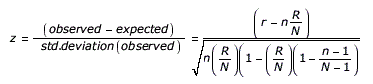
n = All genes associated with a given element
r = Alternatively regulated genes associated with a given element
N = All genes examined
R = All alternatively regulated genes
Once z-scores have been calculated for all protein features and miR-BS linked to alternatively regulated probe sets, a permutation analysis is performed to determine the likelihood of observing these z-scores by chance. This is done by randomly selecting the same number of regulated probe sets from all probe sets examined and recalculating z-scores for all terms 2000 times. The likelihood of a z-score occurring by chance is calculated as the number of times a permutation z-score is greater than or equal to the original z-score divided by 2000. A Benjamini-Hochberg correction is used to transform this p-value to adjust for multiple hypothesis testing.
Gene Ontology and Pathway Over-Representation Analysis
To perform advanced pathway and Gene Ontology (GO) over-representation AltAnalyze includes core python modules from the program GO-Elite (version 1.2). GO-Elite performs Gene Ontology (GO) and WikiPathway over-representation using the same algorithms listed for domain/miR-BS over-representation analysis (ORA) (e.g., z-score, permutation p-value, BH p-value calculation). After ORA, all GO terms and pathways are filtered using user-defined statistical cut-offs (z-score, permutation p-value and number of genes changed), with GO terms further pruned to identify a minimally redundant set of reported GO terms, based on hierarchical relationships and ORA scores (see GO-Elite documentation). The gene database used for GO-Elite is specific to the version of Ensembl in the downloaded AltAnalyze gene database (Plus versions only with associations directly from Affymetrix included). All result files normally produced by GO-Elite will be produced through AltAnalyze.
AltAnalyze generates two types of gene lists for automated analysis in GO-Elite; differentially expressed genes and alternatively regulated genes. Criterion for differentially expressed genes are defined by the user in the GO-Elite parameters window (e.g., fold difference > 2 and t-test p < 0.05). The user can choose to use the rawp (one-way ANOVA, two groups) or adjp (BH adjusted value of the rawp) in the software. All genes associated with alternative exons are used for GO-Elite analysis. Genes with alternative exons that also have alternative splicing annotations can be further selected using the “Filter results for predicted AS” option. Results are exported to the “GO-Elite” directory in the user-defined output directory, while input gene lists can be found in the folders “GO-Elite/input” and “GO-Elite/denominator”. The gene lists for differentially expressed genes have the prefix “GE.” while the alternative exon files have the prefix “AS.”. The appropriate denominator gene files for each is selected by GO-Elite. Species and array specific databases for GO-Elite analysis are downloaded automatically from AltAnalyze when the species database is installed. Array types supported for each species include any Affymetrix supported at the time the Ensembl database was released along with any arrays supported by Ensembl.
Probe Set Filtering
Prior to an Affymetrix alternative exon analysis, AltAnalyze can be used to remove probe sets that are not deemed as sufficiently expressed. For the two conditions that AltAnalyze compares (e.g., cancer versus normal), a probe set will be removed if neither condition has a mean detection above background (DABG) p value less than the user threshold (e.g., 0.05). Likewise, if neither condition has a mean probe set intensity greater than the user threshold (e.g., 70), then the probe set will be excluded from the analysis. When comparing two conditions (pairwise comparison) for probe sets used to determine gene transcription (e.g., constitutive aligning), both conditions will be required to meet these expression thresholds in order to ensure that the genes are expressed in both conditions and thus reliable for detecting alternative exons as opposed to changes in transcription. When comparing all biological groups in the user dataset, however, these additional filters are not used. For RNA-Seq analyses, the read counts associated with each exon or junction are used as a surrogate for the expression. These same filters applied to constitutive annotated exons or junctions and junctions used for reciprocal junction analysis.
Constitutive and Gene Expression Calculation
Identifying exons and junctions for gene expression calculations
A reliable estimate of transcriptional activity for each gene is required to both report basic gene expression statistics and perform alternative exon analysis. Affymetrix exon and junction arrays probe most well annotated exon regions, many introns, untranslated regions and theoretically transcribed regions. Gene expression values can be calculated in one of two ways from AltAnalyze: (A) using features aligned to constitutive exons, or (B) using features aligned to all known exons. Using constitutive exons are recommended for use for all analyses.
Constitutive exons in AltAnalyze are classified as exons that align to a pre-determined set of exon regions that are most common to all mRNA transcripts used when the AltAnalyze database is created. RNA transcript exon structures and genomic positions are extracted from analogous versions of Ensembl and the UCSC genome browser database. While AltAnalyze database versions EnsMart49 and EnsMart52 used mRNA annotations from the Affymetrix probe set annotation file, EnsMart54 and later exclusively derives constitutive exon annotation from UCSC and Ensembl exon structures (Figure 3.1). If only one exon region is considered constitutive then the grouped exon regions with the second highest ranking (based on number of associated transcripts) are included as constitutive (when there at least 3 ranks). For next generation Affymetrix junction arrays (hGlue, HJAY, HTA2.0 and MJAY), constitutive exons are obtained as described above. Constitutive junctions from these arrays are currently determined by two methods: 1) determine if both of the exons in the junction are annotated as constitutive and 2) determine if the junction is annotated as a putative ‘alternative’ probe set according to the manufacturer annotation file (e.g., mjay.r2.annotation_map). If both lines of evidence indicate the probeset is constitutive, then the probeset is annotated accordingly. Probe set annotations are provided in “AltDatabase/EnsMart55/Hs/junction/Hs_Ensembl_probesets.txt” (modify for your species, Ensembl version and array type). For RNA-Seq analyses, junctions are considered constitutive if the 5’ and 3’ splice sites of the detected junction map to the corresponding splice sites of two exons that are; 1) annotated as constitutive and 2) not annotated as alternative from the transcript reciprocal junction comparison analysis. Constitutive RNA-Seq junctions are only evaluated when no exon reads are present.
In addition to constitutive exons, users have the option to use all features that have an Affymetrix core probe set annotation or align to an AltAnalyze defined exon region. Affymetrix defines probe sets according to three criterion, "core", "extended" and "full". The Affymetrix core annotations are recommended for calculation of constitutive levels by Affymetrix, however, these are often a mix of known alternative exons and AltAnalyze annotated constitutive exons. In addition to these Affymetrix core exons, the AltAnalyze core set consists of any probe set that aligns to an exon region, but excludes probe sets aligning to any non-exon features, including introns and untranslated regions (Figure 3.1). In version 2.0 of AltAnalyze, users are no longer restricted to genes containing constitutive annotated exons/junctions for alternative exon analysis. Prior to version 2.0, AltAnalyze removed genes during the filtering process that had no exon or junctions with constitutive annotation prior to the alternative exon analysis. To address this issue, all exon or junction features that align to known mRNAs are used to assess gene expression when no constitutive features are deemed “expressed” according to the user’s filtering parameters.
Calculating gene expression values
Gene expression values are calculated twice during an exon array analysis: (1) To report gene expression fold changes and summary statistics, and (2) To calculate gene expression normalized intensities for alternative exons. While both analyses use the same set of starting exons, junctions or probe sets (constitutive or mRNA aligning), they differ in how filtering of these features occurs. These differences are important when trying to eliminate false positive alternative exon predictions.
For gene expression summary reporting, expression values from all constitutive or core features (user defined) are filtered and then averaged. For this analysis, the constitutive or core features are first identified from the AltAnalyze database (Ensembl_probeset or Ensembl_exon file) and corresponding expression and detection probabilities extracted from the APT RMA result files from the input array files. For RNA-Seq analyses only total number of counts associated with each exon/junction are considered. For each biological group (typically containing replicates), the mean expression and mean detection above background probabilities are calculated for each feature. If the mean expression and the mean detection above background probabilities for at least one group does not meet the user defined thresholds (by default p<0.05 and expression>70), then this feature will not be used to calculate the gene expression value for the corresponding gene. The average expression for all remaining features for each sample will be calculated to obtain a mean gene expression value for each feature for each replicate. Thus, any differences between features used to calculate gene expression will be averaged. For example, when all core features are used, the expression of any alternative exons will be averaged with the non-alternative exons. If no constitutive or core features for a gene meet these criterion (see below), then all will be used to calculate gene expression. Only one set of gene expression values is reported for each Ensembl gene.
This same strategy is used for calculating a gene expression value for the alternative exon analysis with the following differences: (1) a feature used to calculate gene expression must demonstrate evidence expression (expression and detection probability filtering) in both conditions (pairwise comparisons only) or it is removed and (2) if no features remain after expression and detection probability filtering, then features for the gene will NOT be analyzed for the alternative exon analysis. If all groups are being compared for the alternative exon analysis, rather than direct comparison of two groups, then only one biological group must meet the expression and detection probability thresholds for the gene expression (same as for gene expression summary reporting, above). These additional requirements ensure that the gene is expressed at sufficient levels to assess alternative splicing in both compared biological groups. This is important in eliminating false positive changes in feature expression that can occur when the gene is expressed in only one condition.
Alternative Splicing Prediction
To predict whether or not a single feature (EP) or reciprocal junction-pair (JP) associates with an alternative splicing or alternative promoter sequence, AltAnalyze uses two strategies: 1) Identify alternative exons/introns based on de novo isoform comparison and 2) Incorporating splicing predictions from UCSC’s “known_alt.txt” file.
De Novo Splicing Prediction and Exon Annotation
In order to identify exons with alternative splicing or promoters, AltAnalyze compares all available gene transcripts from UCSC and Ensembl to look for shared and different exons. To achieve this, all mRNA transcripts from UCSC’s species-specific “mRNA.txt” file that have genomic coordinates aligning to a single Ensembl gene and all Ensembl transcripts from each Ensembl build are extracted. Only UCSC transcripts that have a distinct exon composition from Ensembl transcripts are used in this analysis, excluding those that have a distinct genomic start or stop position for the first and last exon respectively (typically differing 5’ and 3’ UTR agreement), but identical exon-structure.
When assessing alternative splicing, cases of intron retention are identified first. These regions consist of a single exon that spans an two adjacent exons at least one another transcript for that same gene. These retained introns are stored for later analysis, but eliminated as annotated exons. Remaining exons are clustered based on whether their genomic positions overlap (e.g., alternate 5’ or 3’ start sites). Each exon cluster is considered an exon block with one or more regions, where each block and region is assigned a numerical ID based on genomic rank (e.g., E1.1, E1.2, E2.1, E3.1). For each exon in a transcript, the exon is annotated as corresponding to an exon block and region number (Figure 3.3). All possible pairwise transcript comparisons for each gene are then performed to identify exon pairs that show evidence of alternative exon-cassettes, alternative 3’ or 5’ splice sites or alternative-N or -C terminal exons (Figure 3.3). All transcript exon pairs are considered except for those adjacent to a retained intron. This analysis is performed by comparing the exon block ID and region IDs of an exon and it’s neighboring exons to the exon blocks and regions in the compared transcript. Ultimately, a custom heuristic assigns the appropriate annotation based on these transcript comparisons.
Figure 3.3
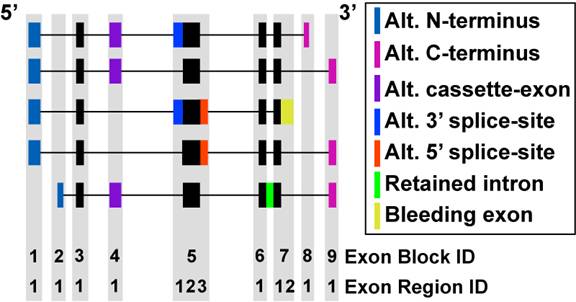
Comparison of mRNA Exon Composition: To determine alternative splicing and alternative promoter regulation, all analyzed transcripts (Ensembl and UCSC) were compared based on exon genomic positions and subsequently annotated by a custom heuristic. Exon block and regions definitions are shown. The different types of alternative exon events are illustrated by different colored exons among five theoretical transcripts for the same gene. The black filled boxes represent exon regions that are most common to all mRNA transcripts. These regions are annotated as constitutive by AltAnalyze. Features that overlap with the constitutive regions can be used to calculate gene expression. All non-intron regions that align to an exon block can also optionally be used to calculate gene expression.
Incorporating UCSC Splicing Predictions
In addition to all de novo splicing annotations, additional splicing annotations are imported from the UCSC genome database and linked to existing exon blocks and regions based on genomic coordinate overlap.This comparison is performed by the alignToKnownAlt.py module of AltAnalyze (called from EnsemblImport.py). De novo and UCSC splicing annotations are stored along with probe set Ensembl gene alignment data in the file \<species>_Ensembl_probesets.txt file (RNA-Seq).
These Affymetrix annotations are used by AltAnalyze and DomainGraph for annotation and visualization. In the AltAnalyze result file, UCSC KnownAlt the major splicing annotation types are altFivePrime, altThreePrime, cassetteExon, altPromoter, bleedingExon and retainedIntron. In comparison, the major AltAnalyze determined splicing annotation types alt-5’, alt-3’, cassette-exon, alt-N-term, intron-retention, exon-region-exclusion and alt-C-term. These splicing annotations are determined by comparing relative exon-junction positions for all analyzed transcripts for each gene (see proceeding section). The annotation "exon-region-exclusion" is the opposite of intron-retention (region most commonly described as exon rather than intron), alt-N-term is similar to altPromoter (distinct 5’ distal-transcript exon with shared 3’ exons) and alt-C-term is the opposite of alt-N-term. To better understand these annotations, look at specific probe set examples through the NetAffx website using the UCSC browser option.
Incorporating Alternative Poly-Adenylation Predictions
Poly-adenylation (polyA) sites are identified using the polyA database version 2. This database provides polyA site genomic coordinates for human, mouse, rat, chicken and zebrafish, generated in 2006 from various genomic database builds. Of these, only human polyA sites are provided as an annotation track in the UCSC genome database. For human, the polyA sites for the appropriate human genome build are downloaded and parsed using the same methods for the knownAlt UCSC annotation track. If multiple polyA sites are observed in a given Ensembl gene, exon regions overlapping with these binding sites are reported as “alternative_polyA” along with the alternative splicing annotations. To report these same annotations for other species, annotations from the polyADB_2 flat-file were converted to UCSC BED format and batch converted to the latest genome build using http://www.genome.ucsc.edu/cgi-bin/hgLiftOver. In some cases, multiple lift-over translations were required. Intermediate files are available at http://www.altanalyze.org/archiveDBs/. Due to missing chromosome annotations for most predictions in zebrafish, these annotations were not included.
Filtering for Alternative Splicing
As mentioned in previous sections, AltAnalyze includes the option restrict alternative exon results to only those probe sets or reciprocal junction-pairs predicted to indicate alternative splicing. AltAnalyze considers alternative splicing as any alternative exon annotation other than an alternative N-terminal exon or alternative promoter annotation derived from de novo or UCSC genome database annotations.
Protein/RNA Inference Analysis
Identifying Alternative Proteins/Protein Domains
RNA-Seq and probe set sequences are used to identify which proteins align to or are missing from transcripts for that gene. To do this, all Ensembl and UCSC mRNA transcripts are extracted for a gene that corresponds to a given feature (probe set or RNA-Seq junction). For each transcript, all exon genomic coordinates are stored. For exon-level analyses, transcripts with exons that contain the probe set (or RNA-Seq exon region) genomic coordinates are considered transcript aligning, while all others are considered non-aligning. For junction analyses, if two reciprocal junctions (or junction and an exon) align to distinct isoforms, these relationships are stored rather than the aligning and non-aligning isoforms, however, if both isoforms do not align to distinct transcripts, then the one with a aligning and non-aligning set of transcripts is stored for further exon comparisons.
If a set of aligning and non-aligning isoforms (competitive) is identified for a single feature or reciprocal junction pair, all possible aligning and non-aligning pairwise combinations are identified to find those pairwise comparisons with the smallest difference in exon composition. This is accomplished by determining the number of different and common exons each transcript pair contains (based on genomic start and stop of the exon). When comparing the different transcript pairs, the most optimal pair is selected by first considering the combined number of distinct exons in both transcripts and second the number of common transcripts. Thus, if one transcript pair has 4 exons in common and 2 exons not in common, while a second pair has 5 exons in common and 3 exon not in common, the prior will be selected as the optimal since it contains less overall differences in exon composition (even though it has less common exons than the other pair). A theoretical example is illustrated in Figure 3.4.
Figure 3.4
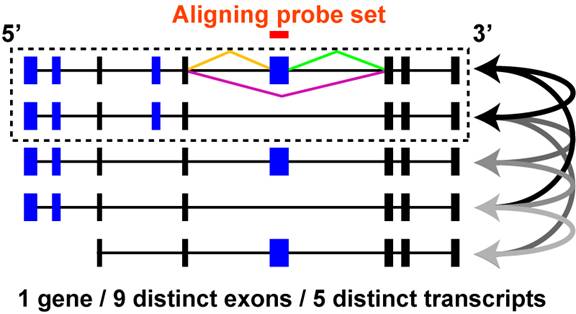
Comparison of probe set aligning and non-aligning mRNAs: A theoretical gene is shown with 9 distinct exons and 5 distinct mRNA transcripts with different exon combinations. Four exons are alternatively expressed in different transcripts (blue exons) while five are common or constitutive to all transcripts (black exons). All possible pairwise transcript combinations are shown (arrows) between mRNAs that contain a probe set aligning exon and those that do not. Ultimately, a single pair is selected that has the most common exons and the least uncommon exons (dashed box). The same strategy is used for junction analyses as exon, however, for junction sensitive platforms, aligning transcripts are identified by direct junction (orange, green, purple) sequence alignment to the mRNAs prior to comparing exon composition as described above.
Once a single optimal isoform pair has been identified, protein sequence is obtained for each by identifying protein IDs that correspond to the mRNA (Ensembl or NCBI) and if not available, a predicted protein sequence is derived based on in silico translation. Although such a protein sequence may not be valid, given that translation of the protein may not occur, these sequences provide AltAnalyze the basis for identifying conservative changes predictions for a change in protein size, sequence and domain composition. Domain/protein features are obtained directly from UniProt’s sequence annotation features or from Ensembl’s InterPro sequence annotations (alignment e-value <1) (see section 5 data extraction protocols). Any InterPro sequences with a description field or any UniProt sequence annotation feature that is not of the type “CHAIN”, “CONFLICT”, “VARIANT”, “VARSPLIC” and “VAR_SEQ” are examined by AltAnalyze. To compare domain or motif sequence composition differences, the protein sequence that corresponds to the amino-acid start and stop positions of a domain for each transcript is searched for in each of the compared protein isoforms. If the length of a motif sequence is less than 6 amino-acids, flanking sequence is included. If a domain is present in one but not another isoform, that domain is stored as differentially present. To identify differences in protein sequence (e.g., alternative-N-terminus, C-terminus, truncation coding sequence and protein length), the two protein sequences are directly compared for shared sequence in the first and last five residues and comparison of the entire sequences. If the N-terminal sequence is common to both isoforms but there is a reduction in more than 50% of the sequence length, the comparison is annotated as truncated. If an Ensembl transcript, non-coding and nonsense mediated decay annotations are included from the Ensembl translation annotations. All of these annotations are stored for each junction, exon or probe set for import into AltAnalyze, with each new database build.
Direct Domain/Motif Genomic Alignment
The above strategy allows AltAnalyze to identify predicted protein domains and motifs that are found in one isoform but not the other (aligning to a feature and not aligning). In addition to these “inferred” domain predictions, that include protein domains/motifs that do not necessarily overlap with the regulated feature, AltAnalyze includes a distinct set of annotations that only corresponds to domain/motif protein sequence that directly overlaps with a feature. Alignment of features to InterPro IDs is achieved by comparing feature genomic coordinates to InterPro genomic coordinates. To obtain InterPro genomic start and end positions are determined by first identifying the relative amino-acid positions of an InterPro region in an Ensembl protein, finding which exon and at what position the InterPro region begins and ends and finally stroing the genomic position of these relative exon coordinates.
These feature-InterPro overlaps can be of two types: 1) features whose sequence is present in the domain coding RNA sequence and 2) features whose sequence is not present in the domain coding RNA sequence. The second type of overlap typically occurs in the gene introns. While these associations are typically meaningful, false “indirect” associations are possible. To reduce the occurrences of these false positives, any feature that aligns to the UTR of an Ensembl gene or that occurs in the first or last exon of an mRNA transcript are excluded. These heuristics were chosen after looking at specific examples that the authors considered to be potential false positives. For junction analyses (RNA-Seq and junction array), the associated critical exon genomic position rather than probe set is used.
Identifying microRNA Binding Sites associated with Alternative Exons
MicroRNA binding site (miR-BS) sequence is obtained and compared from five different microRNA databases (see Extracting microRNA Binding Annotations Overview), and compared to identify miR-BSs in common to or distinct to different databases. Probe set sequences are obtained from Affymetrix, while critical exon sequences (corresponding to two reciprocal junctions) are obtained from chromosome FASTA sequence files (see Required Files for Manual Update). Each miR-BS sequence is searched for within probe set or critical exon sequence to identify a match. These relationships are stored with each new database build and are used by AltAnalyze and DomainGraph.
Exhaustive Protein Domain/Motif Analysis
Very similar to the competitive protein domain/motif analysis is the exhaustive protein comparison analysis. This feature is currently not available by default in AltAnalyze and requires replacement database files from AltAnalyze support. The purpose of these files is to obtain the most conservative possible domain-level prediction results from the competitive analysis. This done by storing all pairwise aligning and non-aligning isoform comparisons (Figure 3.4) and then obtaining protein sequence for each transcript, as described in earlier sections and storing all domain/motifs differentially found between all possible competitive isoforms. From these stored results, all possible competitive isoforms are themselves compared to find isoforms that ideally show only differences in central regions of the protein (no N-terminal or C-terminal differences), next for those that contain as few possible domain-level predictions and finally for those with the smallest overall differences in protein length. For detailed algorithm information see the IdentifyAltIsoforms module and the function “compareProteinFeaturesForPairwiseComps”.
Gene Annotation Assignment
AltAnalyze extracts useful gene annotations for determining whether a gene (A) encodes for protein, (B) is likely “drug-able”, (C) is a transcriptional regulator, (D) has putative microRNA targets, (E) participates in known pathways, (F) localizes to specific cellular compartment and (G) is a housekeeping gene. To obtain these annotations, AltAnalyze extracts various annotations for several public genomic databases (Ensembl, UniProt, miRBase, TargetScan, miRanda, Pictar, miRNAMap, WikiPathways, Gene Ontology, Affymetrix). Extracted from Ensembl are annotations for protein coding potential in addition to pseudogenes, rRNA, miRNAs, snoRNAs, lincRNA and other gene types. From UniProt, annotations for kinase, GPCR and coupling pathway, single transmembrane receptors transcriptional regulator and cellular compartment. Annotations for Gene Ontology termas and WikiPathways are extracted on the fly from the downloaded Official GO-Elite annotation databases. Housekeeping gene predictions are extracted from this same database, by searching for Ensembl-Affymetrix associations for probesets containing the ‘AFFX’ notation. Any microRNA targeting predictions from miRBase, TargetScan, miRanda, Pictar, miRNAMap (see Extracting microRNA Binding Annotations Overview) are reported along with the source of the annotation and overlapping predictions.